内容
概述
这个ReactomeFIViz应用程序的目的是找到与癌症和其他类型疾病相关的途径和网络模式。这个应用程序访问反应途径存储在数据库中的通路,帮助您对一组基因进行通路富集分析,直接在Cytoscape中使用手工绘制的通路图可视化hit pathway,并研究hit pathway中基因之间的功能关系。应用程序还可以访问Reactome功能交互(FI)网络,一个高度可靠,手动策划pathway-based蛋白质功能交互网络覆盖超过60%的人类蛋白质,并允许您构建一个FI子网络基于一组基因,查询FI为底层数据源交互的证据,构建分析高相互作用基因群网络模块,进行功能富集分析对模块进行注释,通过查找与实验数据集相关的基因扩展网络,显示路径图,叠加癌症基因索引注释等多种信息源。最近,我们还增加了一些功能,帮助用户在FI网络和Reactome途径的背景下可视化fda批准的癌症药物,并使用直接从Reactome途径构建的布尔网络模型来调查显示的癌症药物的潜在功能影响。
例如,我们如何使用Reactome FIs进行癌症数据分析,请参阅我们的出版物:人类功能蛋白相互作用网络及其在癌症数据分析中的应用.
下载并启动ReactomeFIViz
ReactomeFIViz app 6需要Cytoscape 3.7.0或以上版本。如果你没有安装Cytoscape 3.7.0或以上版本,请从Cytoscape的网站下载:http://www.cytoscape.org. 启动Cytoscape后,使用菜单“应用程序/应用程序管理器”打开“应用程序管理器”对话框,并搜索“ReactomeFI”。您应该看到中间面板中列出的RealToMeViz应用程序(参见下图),您可能会看到不同的版本号。注意:此应用程序的列出的名称是“ReactomeFipugin”,这是该应用程序的原始名称)。选择应用程序,然后点击对话框底部的“安装”按钮。按照步骤完成安装。
使用反应途径
使用途径可视化和分析功能,可以将反应数据库中的路径加载到Cytoscape中,在本机途径图视图或FI网络视图中可视化反应态途径,对一组基因进行途径浓缩分析,以及从您的检查基因列表在点击路线中。
探索Reactome通路
- Load Reactome pathways:使用“Apps/Reactome FI/Reactome pathways”菜单将路径加载到Cytoscape中。加载的路径按照层级方式组织,就像Reactome web应用程序(https://reacectome.org/pathwaybrowser/),并在名为“Reactome”选项卡的左侧“控制面板”中列出。
 反应途径
反应途径 - 在Reactome中查看路径:在路径层级中选择路径后,可以从弹出菜单中选择“查看Reactome源”(在Windows中单击右键,或在mac中单击control,得到弹出菜单)在反应中查看其详细注释。或者,您可以选择“反垃圾中的视图”,以查看反垃圾邮件应用程序中的详细信息。
 路径弹出菜单
路径弹出菜单
请注意:所选路径的祖先路径(容器路径)显示在中间面板的Reactome选项卡中的“selected Event Branch”中。您可以在中间面板中单击一个祖先路径,以查看单击的路径在原始路径层次树中的位置。然而,祖先路径将不会在原始树中被选择。这是一种将选择保存在原始树中的设计行为。 - 搜索路径:在弹出菜单中选择“搜索”以打开搜索对话框。找到的路径将在路径树中以蓝色突出显示。
请注意:搜索将针对所有加载的路径,不限于所选路径及其包含的子路径。 搜索路径
搜索路径 - 开放的reacome Reacfoam: Reacfoam视图提供了reacome数据库中所有(不包括疾病)人类路径的整体视图。在弹出菜单中选择“打开Reactome Reacfoam”,在默认浏览器中打开Reactome Reacfoam。
请注意:按下鼠标并将其按住路径框,将自动选择ReactomeFIV树中的路径。 开放式泡沫
开放式泡沫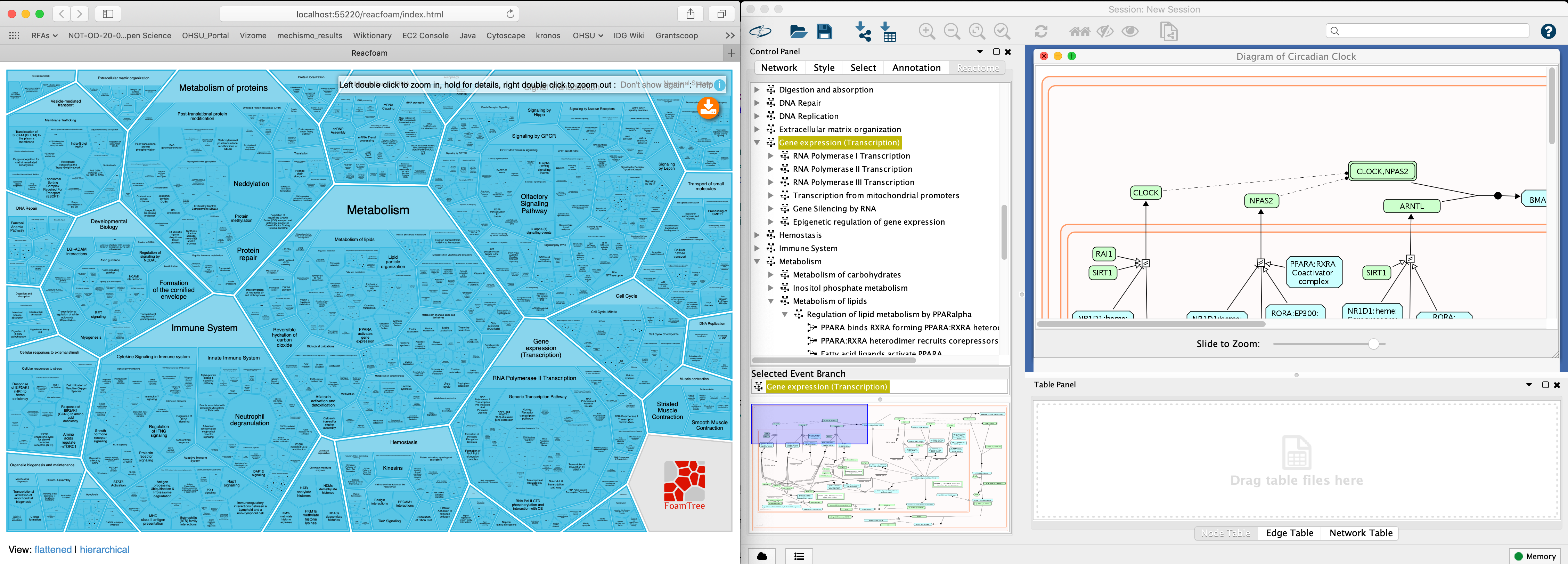 反应reacfoam
反应reacfoam - 开放路径图:Reactome中的路径以分层的方式组织。不是所有的路径都有自己的路径图。一个更小的路径(称为子路径)可以在一个更大的路径中绘制,后者有自己的路径图。大多数顶级通路(称为模块或超级通路)用于组织相关通路(如疾病、信号转导),因此只包含代表规范通路的矩形框。
- 显示图表:如果选定的路径有其自己的通路图,可以在弹出菜单中选择“显示图表”,以将其路径图打开到中央Cytoscape桌面。
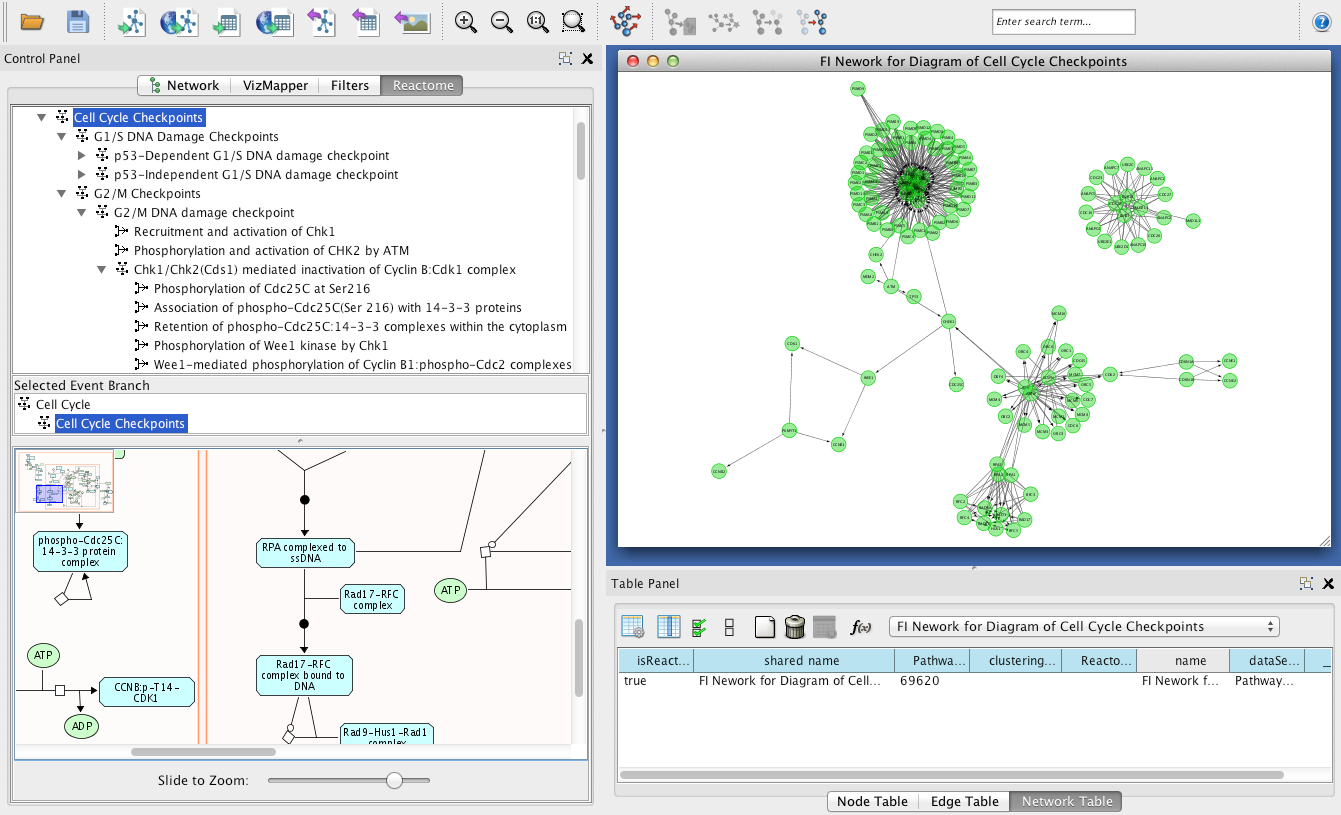
- 在图:如果所选路径被布置为较大路径中的子路径,则可以在弹出菜单中选择“在图表中查看”,以在其容器路径中查看其图形。打开图表后,所选路径包含的反应将以蓝色突出显示。例如,参见下面路径“细胞周期检查点”中打开的路径“G1/S DNA损伤检查点”:
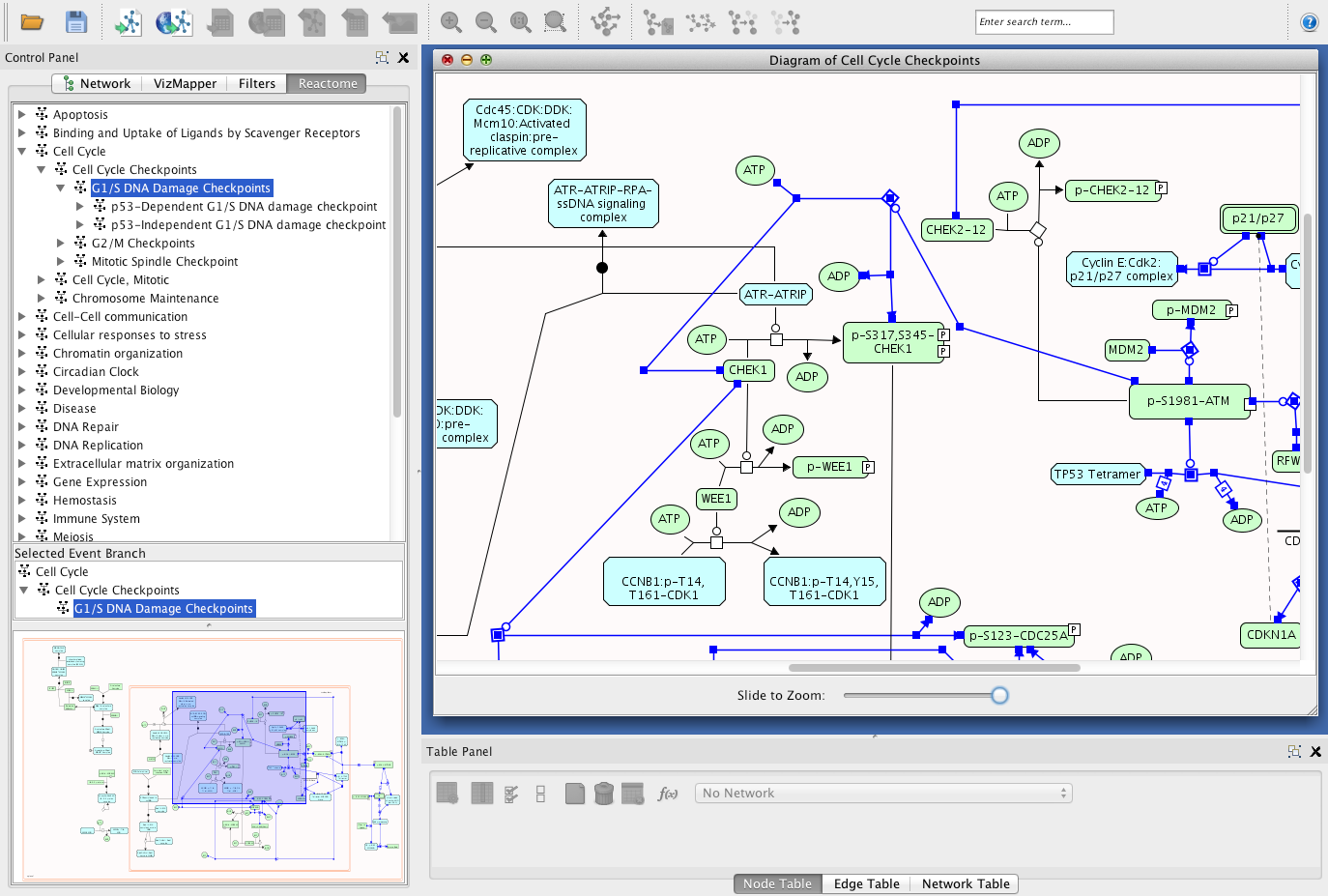 通路图
通路图
- 搜索图:路径图中显示的对象可以在弹出菜单中使用“搜索图”进行搜索(在Windows中单击右键,或者在mac中不选择路径图中的任何对象而单击Control,即可得到弹出菜单)。找到的对象将被选中并以蓝色高亮显示。
请注意:在图中不会搜索反应。使用路径树中的搜索特性来搜索反应。 - 导出图:可导出为PDF、JPG、PNG格式。使用弹出菜单中的“导出关系图”导出显示的关系图。
- 查看Reactome源或在Reactome中查看:选择一个对象,然后右键单击(或控制单击)获得弹出菜单。选择“View Reactome Source”查看表中所选对象的详细注释(示例见下图)。或者选择“在Reactome中查看”,在Reactome web应用程序中查看所选对象。
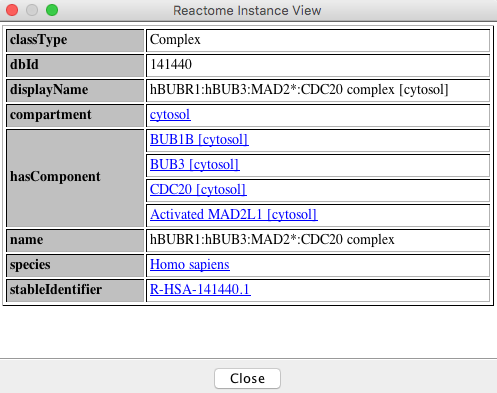 Reactome实例视图
Reactome实例视图 - 列表基因:在选择一个对象后,可以使用菜单项“列表基因”查看一个复杂或蛋白质组所包含的基因,或与显示的蛋白质相关的基因。例如,下面的对话框显示了复杂的hBUBR1:hBUB3:MAD2*:CDC20所包含的基因。点击一个基因符号将带你到GeneCard网站的基因网页。
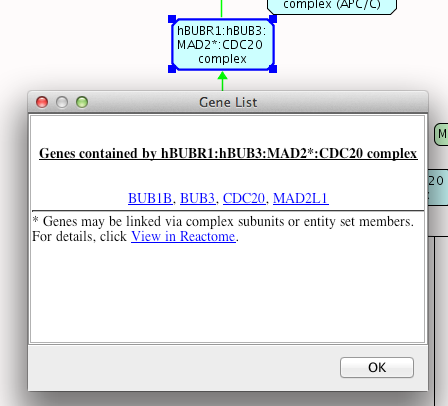 基因列表
基因列表








在FI网络视图中显示反应路径
- 在FI网络视图中显示路径:一个Reactome路径可以通过我们建立的方法转换成一个功能性的相互作用网络(见人功能蛋白相互作用网络及其在癌症数据分析中的应用).在弹出菜单中使用“转换为FI网络”,通过右键单击(窗口)或控制按钮单击(MAC)在路径图面板中没有任何选择的空白区域。原始路径图将移动到左下角,并根据原始路径图生成新的FI网络,该网络将显示在新的网络面板中。
请注意:显示路径包含的子路径也将被提取到FI网络中。
 FI网络视图中的路径
FI网络视图中的路径 - 探索路径和网络视图中的对象:已同步三个视图中的对象选择。可以选择的对象包括:路径树视图中的事件、左下角路径视图中的对象以及网络视图中的基因和FI。您可以在三个视图中的一个视图中选择一个对象,也可以在其他两个视图中选择相应的对象。此外,您还应该在每个视图中使用弹出菜单中实现的功能,以便像在单个视图中一样浏览对象。

请注意:使用Cytoscape的内置“保存会话”功能可以从路径中保存转换的FI网络。但是,显示的路径无法暂时保存到会话文件中。我们将在将来的释放中实施此功能。
通路富集分析
- 通路富集分析:基因列表可以用来检查是否有任何的Reactome途径被富集。为此,使用弹出菜单项“Analyze Pathway Enrichment”(左下方图),获得选择基因集文件的对话框(右下方图)。您可以使用三种文件格式之一的基因集文件:每行一个基因,所有基因在同一行并以逗号分隔,或所有基因在同一行并以制表符分隔。你也可以通过点击“点击进入”按钮手动输入基因(一行代表一个基因)。
请注意:依赖于基因列表的大小,可能需要1分钟以运行途径浓缩分析。该特征中使用的途径与用于注释FI网络或网络模块的反应态途径不同。这里所有超过2,000个路径都使用。对于注释,使用已经预先选择的反应型途径的子集进行了一定尺寸。
请注意:要获得途径浓缩分析结果的整体视图,请在分析后使用弹出菜单“打开反垃圾reacfoam”进行途径途径的反应Reacfoam。您可以通过单击右上角的下载按钮下载ReaCFoam视图。要为Windows 10用户打开ReaCFOAM视图,您需要通过在“系统和安全性”控制设置中的“允许应用程序”的设置中“公共”来允许“公共”访问Cytoscape。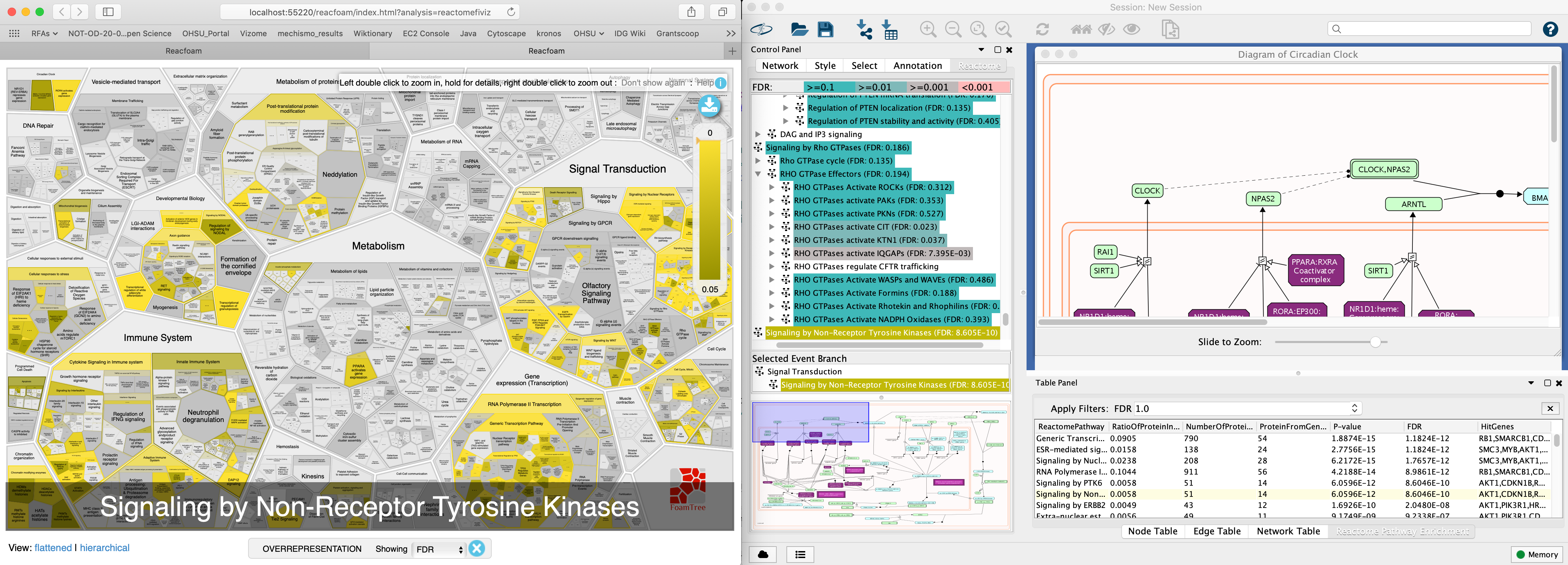
- 查看浓缩分析结果:途径富集结果在Cytoscape主窗口下方的“table Panel”中以“Reactome Pathway enrichment”的表格形式显示。可以使用“图中的视图”在路径图视图中查看命中路径,使用“导出标注”将结果保存到表中。Reactome路径树中的路径根据它们的FDR值以不同的颜色突出显示。在路径图视图中,包含基因列表中的基因的对象以紫色背景和白色字体高亮显示。在FI网络视图中,命中路径的命中基因显示在紫色的粗边上。
请注意:在“基因列表”功能的“基因列表”对话框中,命中基因以相同的颜色显示。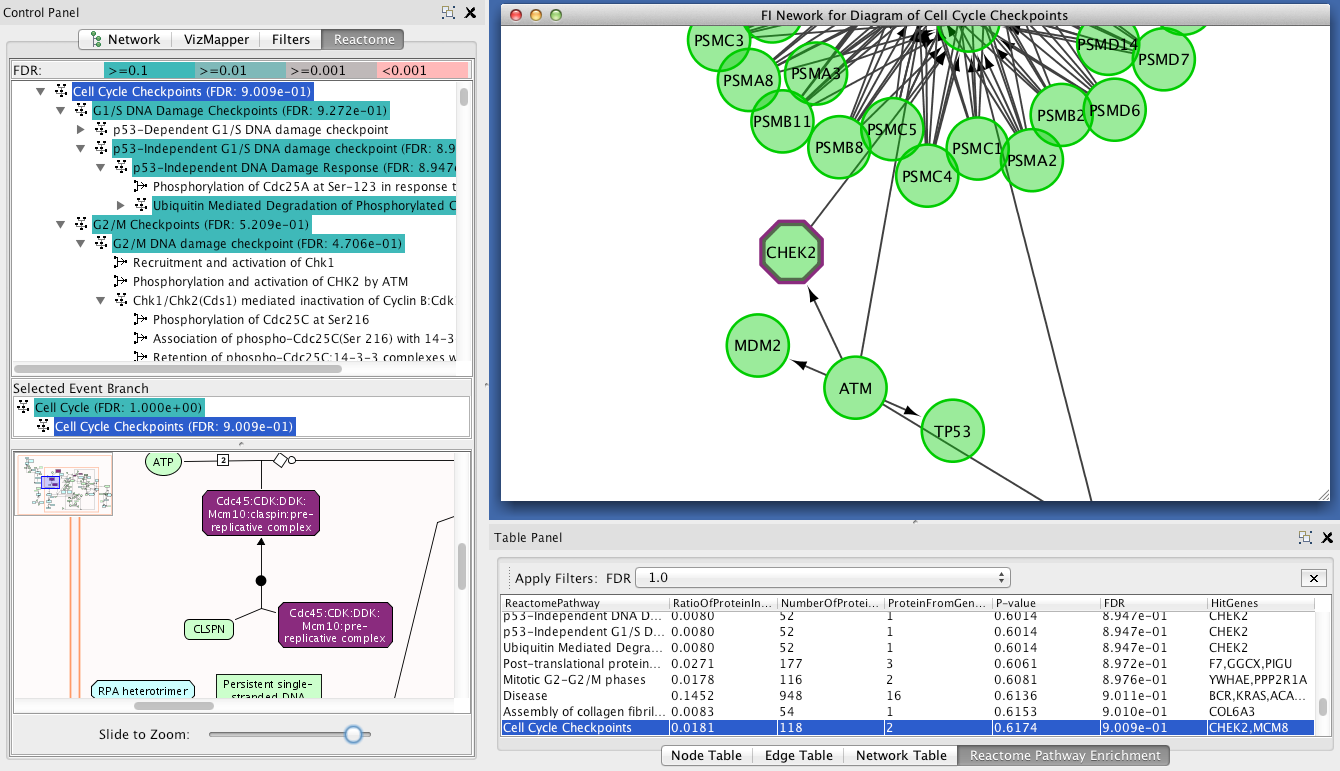 通路富集的结果
通路富集的结果 - 执行GSEA分析:基因集浓缩分析(GSEA)是一种基于秩的路径富集分析方法,广泛应用于基于路径的数据分析。ReactomeFIViz支持使用基因评分文件对Reactome路径进行GSEA分析。基因评分可以是来自差异基因表达分析的t评分,也可以是其他类型的评分,可以进行排名。要执行GSEA路径富集分析,需要提供一个以制表符分隔的文本文件,其中包含两列:第一列用于基因符号(仅用于人类),第二列用于基因得分。第一行保留给列标题,不会导入进行分析。要进行GSEA分析,在路径树中使用弹出菜单“perform GSEA analysis”,弹出GSEA配置对话框,在对话框中输入基因评分文件,选择最小和最大路径大小以及排列数。
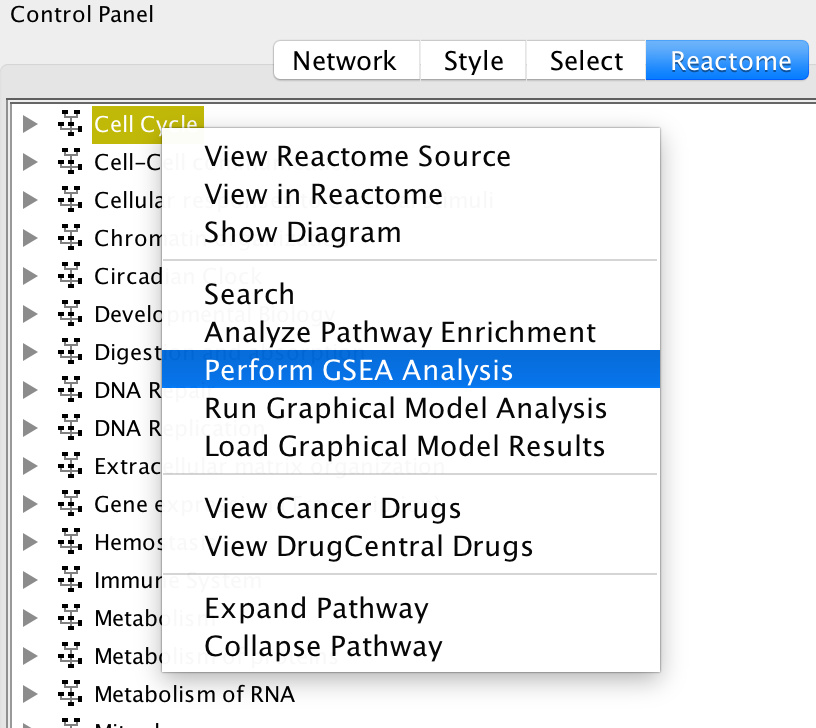 执行GSEA分析
执行GSEA分析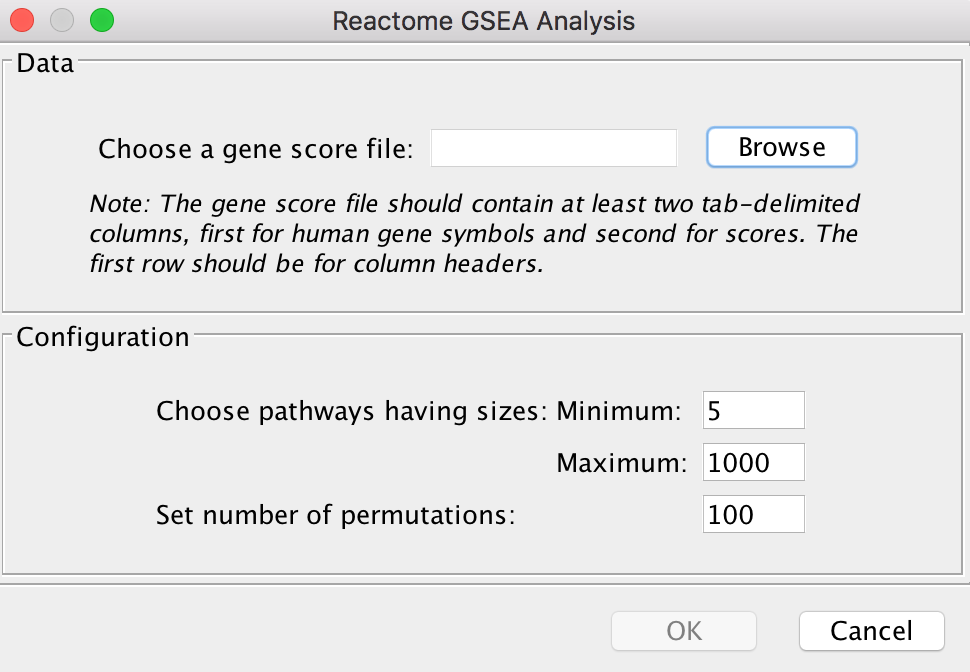 配置GSEA分析
配置GSEA分析GSEA分析结果显示在Cytoscape table Panel中“Reactome GSEA analysis”的表格中。在基于基因集的路径富集分析中,路径树中需要进行GSEA分析的路径根据FDR值突出显示(见上文)。结果表中各列的含义请参考GSEA原始文档:GSEA文档.
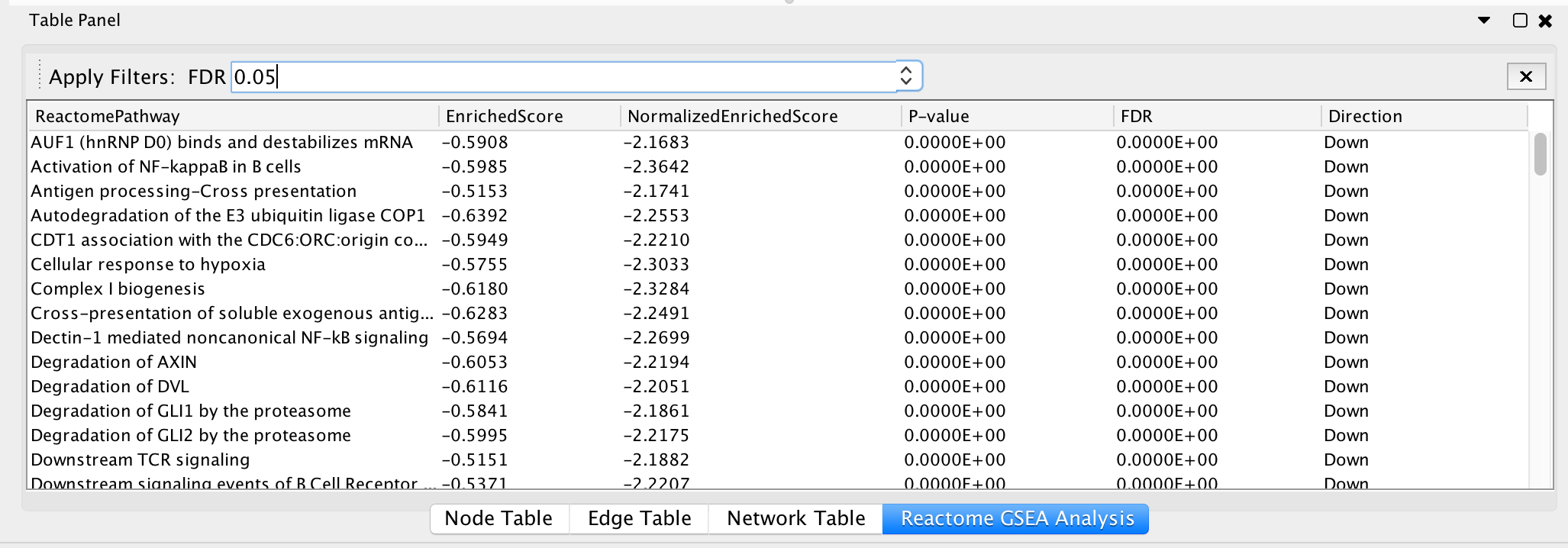 GSEA分析结果
GSEA分析结果 - 将基因分数叠加到路径上:对于GSEA分析产生的重要通路,可以叠加基因评分,以调查具有显著高或低评分的基因产品的位置,从而了解这些极端评分造成的潜在通路活性影响。为此,在路径图视图中使用弹出菜单“覆盖基因评分”,在配置对话框中选择基因评分文件。文件加载后,路径图中的实体将根据分数高亮显示。您可以在正确的基因评分表视图中选择一个或多个基因,以可视化路径图中的相关实体。
 重叠基因的分数
重叠基因的分数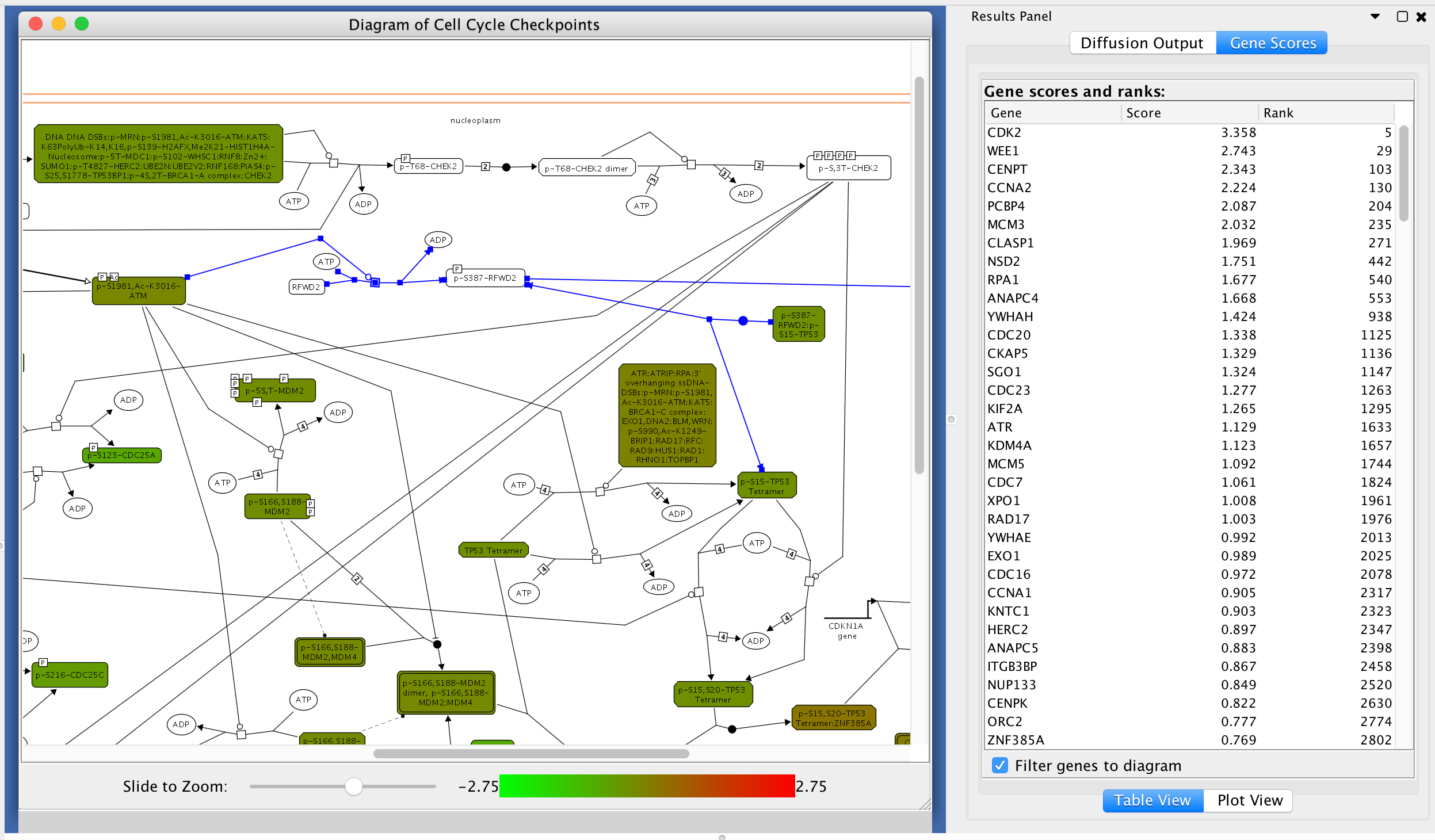 基因评分叠加结果
基因评分叠加结果请注意:如果一个实体(如复合体或实体集)由多个基因组成,则该实体的得分为该实体注释的所有基因的平均值。要删除路径图中重叠的基因分数,请使用弹出菜单“删除基因分数”。要查看显示的路径图中注释的基因的分数分布,请在Cytoscape结果面板的“基因分数”选项卡中选择“绘图视图”(见下文)。
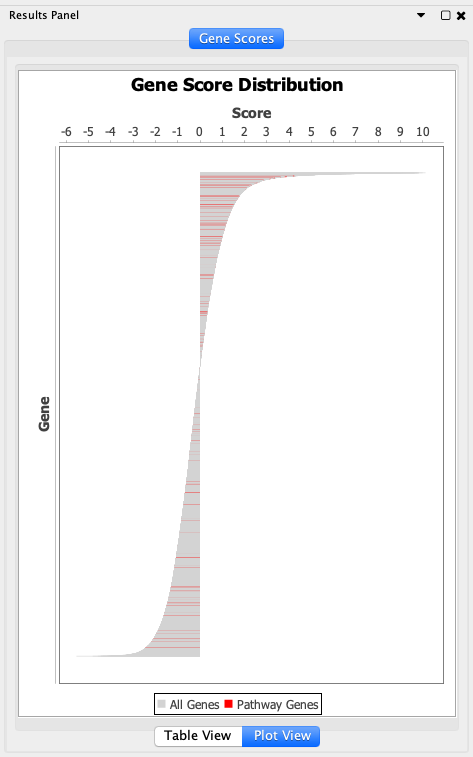 基因评分分布
基因评分分布










基于概率图形模型的路径分析
我们采用了Reactome路径的范式方法,将路径图中绘制的反应转化为因素图中的因素,这是一种概率图形模型(PGMs)。有关PARADIGM方法的详细信息,请参见:利用PARADIGM从多维癌症基因组学数据推断患者特异性通路活动.有关因子图的介绍,请看维基百科的这个条目:因子图. 出于测试目的,您可以下载100名TCGA卵巢癌患者的两个样本数据文件:CNV和信使rna基因表达.原始TCGA OV文件被下载广泛的机构网站。
- 批量运行图形模型分析:此功能用于对具有手工布局图的所有Reactome路径执行批量图形模型分析。
- 开始分析:在路径层次树中选择弹出菜单“运行图形模型分析”。选择此菜单后,在“运行图形模型分析”对话框的以下两个选项卡中将要求您选择数据文件并提供推理算法的参数:
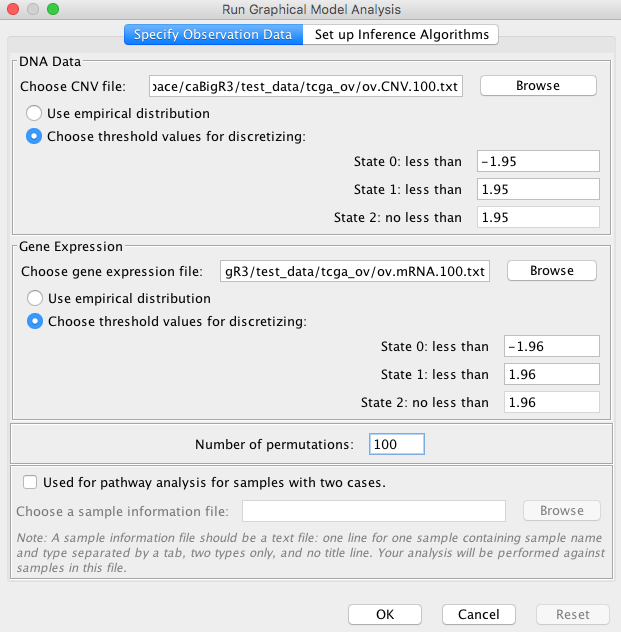 LoadData
LoadData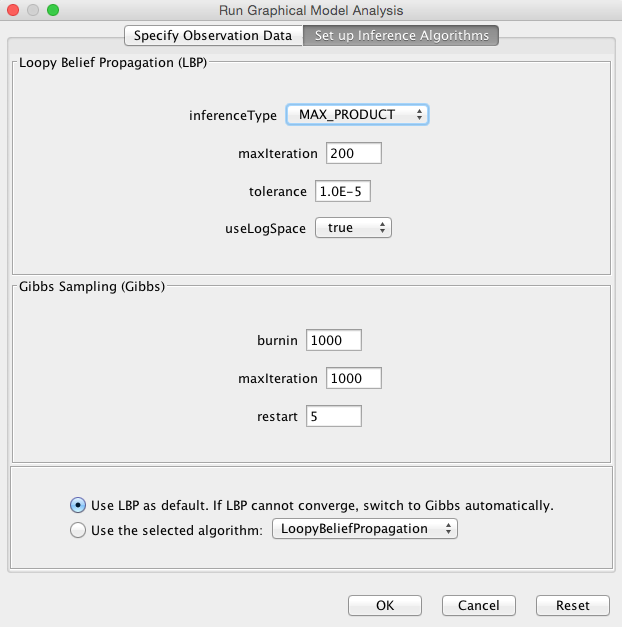 SetUpAlgorithms
SetUpAlgorithms
笔记:
1).如果在数据加载对话框中选择“使用经验分布”,加载的数据将直接用于构建因子函数,而不进行离散化。目前,我们建议采用“选择阈值进行离散化”。
2). 建议使用推理算法的默认参数进行批量分析,以实现快速性能。在从批处理分析中找到有趣的路径后,您可以为某些特定路径尝试不同的参数。如果要执行两个案例研究(例如,病例对照、药物敏感/不敏感等),请选中复选框“用于两个案例样本的路径分析”,并根据需要提供样本信息文件。对于两个案例分析,将不会生成随机数据集。结果将通过比较上传数据文件中的两种样本来显示。 - 运行分析:点击“确定”按钮,开始批量分析。根据你的样本量,完成整个分析可能需要几个小时。
- 完成分析:在批分析完成后,如果由于推理算法不能收敛而无法分析某些路径,您可能会看到以下列表。请确保以下列表很小(可能少于10条路径),这样你就能得到足够的结果。
 FailedPathwaysList
FailedPathwaysList - 查看结果:批分析的结果显示在Cytoscape桌面的底部表格面板上,如下所示:请注意:此表中有7列:ReactOmePath用于应用程序分析的路径名称;AverageUpIPA通过与随机背景进行比较,显示一条通路受到的干扰程度(IPA:综合通路活动。有关详细信息,请参阅上述范例论文);AverageDownIPA显示了一条通路被向下扰动的程度;CombinedPValue是一个p值,表示基于路径输出和Fisher方法,该路径受到干扰的程度;MinimumPValue是路径输出的最小p值;最后两列是基于Benjamini–Hochberg方法的两个p值的FDR。AverageUpIPA或AverageDownIPA可能为NaN,这表示根据此分析未检测到向上或向下扰动。FDR过滤基于最后两列中显示的FDR值,并使用“或”操作。
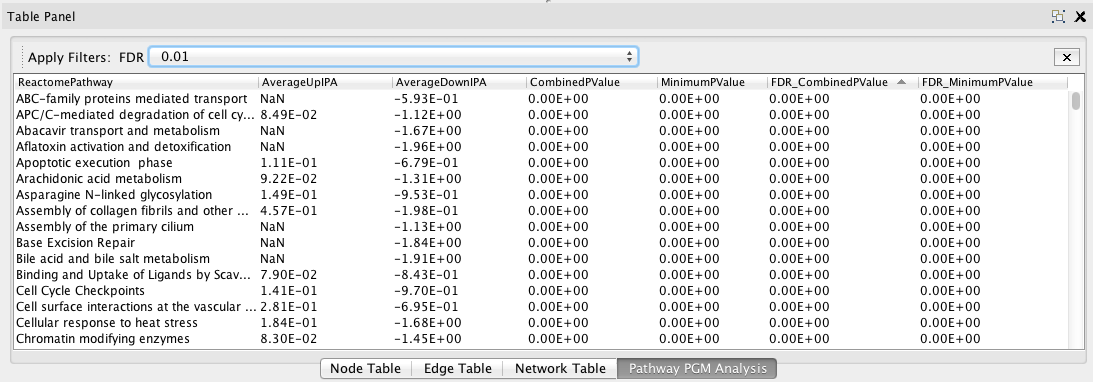 BatchResults
BatchResults - 保存结果:要保持结果,请使用表中的弹出菜单“导出注释”,将结果保存到外部文本文件中。通过使用POPUP菜单,“装载图形模型结果”,在路径树中可以稍后加载已保存的结果。
- 开始分析:在路径层次树中选择弹出菜单“运行图形模型分析”。选择此菜单后,在“运行图形模型分析”对话框的以下两个选项卡中将要求您选择数据文件并提供推理算法的参数:
- 对单个路径进行图形模型分析:此特性用于对显示在Cytoscape桌面中的路径执行图形模型分析。
- 开放的途径:如前所述,您可以在路径树中选择路径,并在Cytoscape桌面中打开其图表。或者,您可以通过选择弹出菜单“查看图表”从批次分析结果表中选择一个有趣的路径。
- 开始分析:从路径图窗口的弹出菜单列表中选择弹出菜单,“运行图形模型分析”。您将被要求提供数据文件,并在批处理分析中设置推理算法。点击“OK”按钮后,你会被要求在下面的对话框中提供路径中不会被图形模型考虑的实体的转义名称列表(如ATP、ADP等):
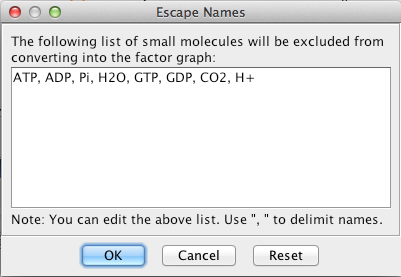 EscapeNameDialog
EscapeNameDialog
请注意:如果已加载数据文件,则可以选择使用加载的数据文件,而不显示“数据加载”对话框。 - 查看结果:分析完成后,Cytoscape的表窗格中将显示三个选项卡:IPA路径分析、IPA样本分析和IPA节点值。IPA路径分析通过比较数据文件中的样本和应用程序基于数据文件动态生成的随机数据集中的样本,显示路径中实体的推断结果。IPA样本分析将每个样本的结果显示为向上或向下扰动。您可以选择显示/隐藏表中样本的p值和FDR值。IPA节点值显示每个样本的路径图中选定实体的推断结果。路径图中的实体根据IPA路径分析选项卡中MeanDiff列中的值高亮显示。
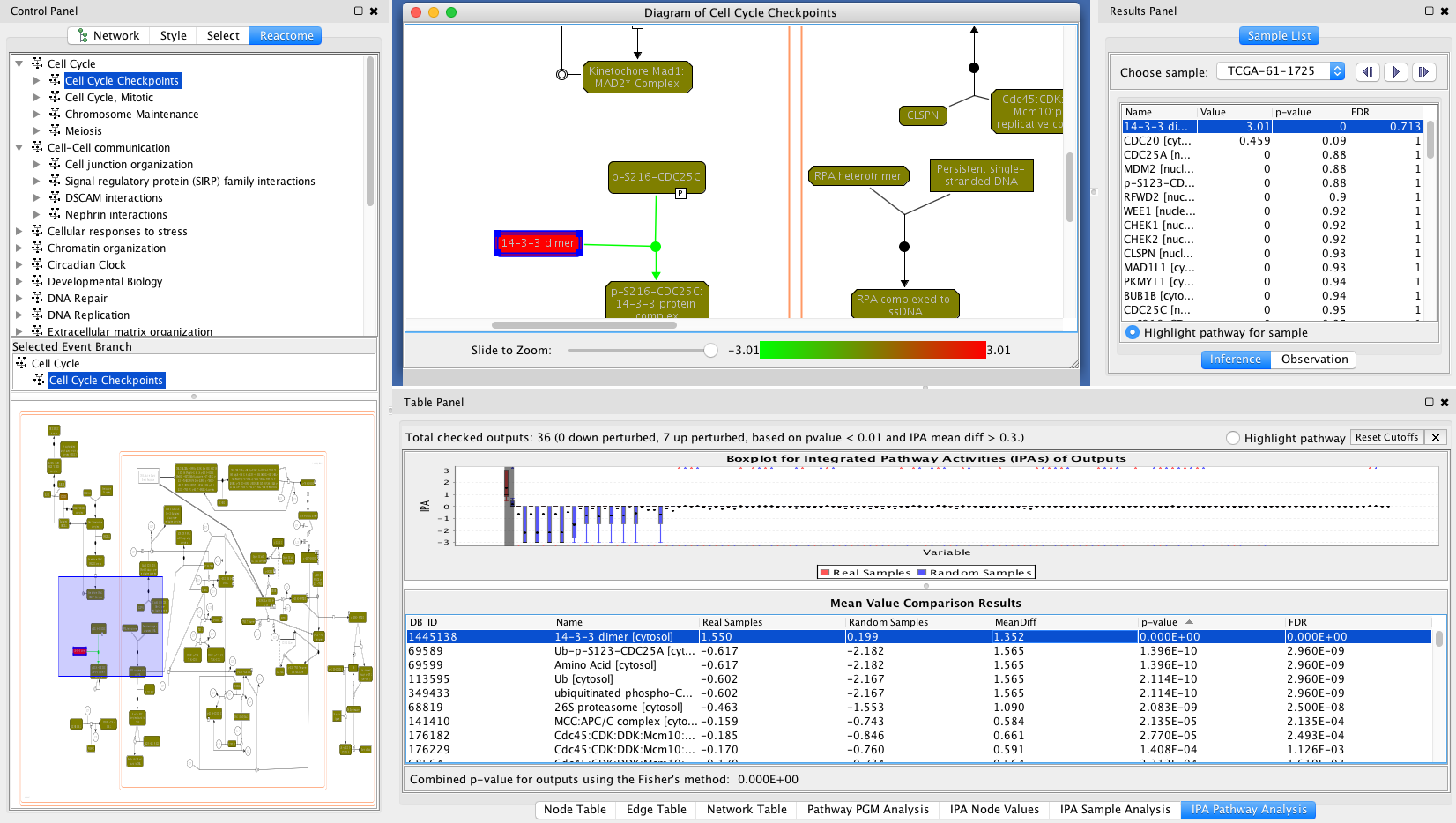 cellcyclecheckpointsresults.
cellcyclecheckpointsresults.
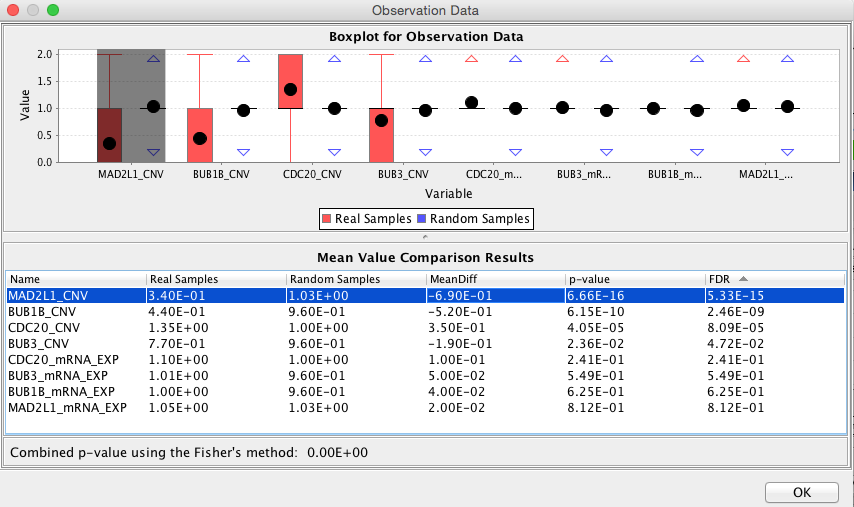
请注意:您可以通过双击路径图窗口底部的颜色频谱栏来更改路径图突出显示的颜色频谱映射,以获取对话框以设置最小/最大值。您可以使用弹出菜单“保存分析结果”,并通过“打开分析结果”稍后将结果加载,并将结果保存分析结果。 - 分析基因水平结果:根据单个基因的基因组数据文件推断出上下扰动结果。该应用程序提供了分析单个基因的观察结果和推断结果的功能。您可以使用弹出菜单“显示基因水平分析结果”和“显示观察结果”,查看整个路径的基因水平观察和推断结果。在选择实体并使用这两个弹出菜单后,您还可以查看路径图中显示的实体包含的基因的这些结果。以下两个对话框显示了基因水平的观察结果和推断结果,这些基因的产物包含在细胞周期检查点途径中的复合物“hBUBR1:hBUB3:MAD2*:CDC20复合物[cytosol]”中:
 ObservationsForEntity
ObservationsForEntity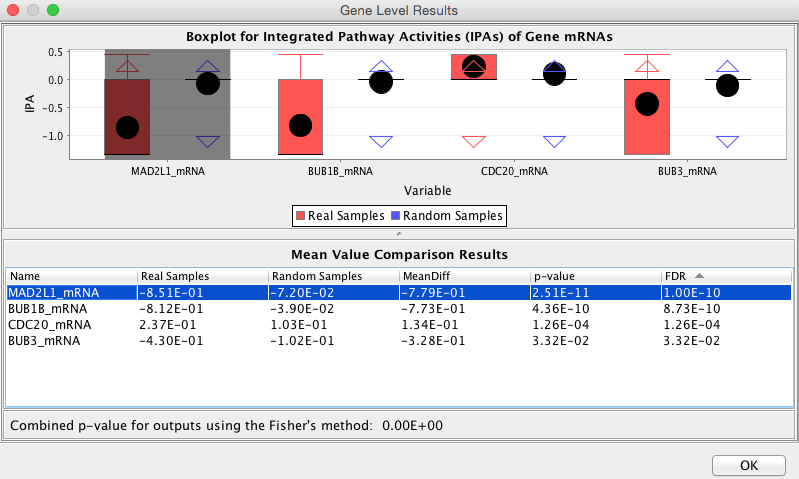 GeneLevelResultsForEntity
GeneLevelResultsForEntity - 分析和可视化个别样本的结果:推论结果和加载的观测数据显示在右边的“结果面板”中(见下)。通过勾选“为样本突出路径”,将根据“选择样本”框中所显示的选定样本的推断IPA值突出显示路径图中的实体。您也可以通过点击播放按钮来启用动画。有两个选项卡“结果面板”:“推理”显示推断出来的价值观,“观察”选项卡加载相关的观测数据通路中的实体(注意:如果你选择“离散化”,显示的观察值是离散:0低于正常水平,为正常,1和2高等师范)。三个视图(路径图、推断表和观察表)中的对象同步进行选择。
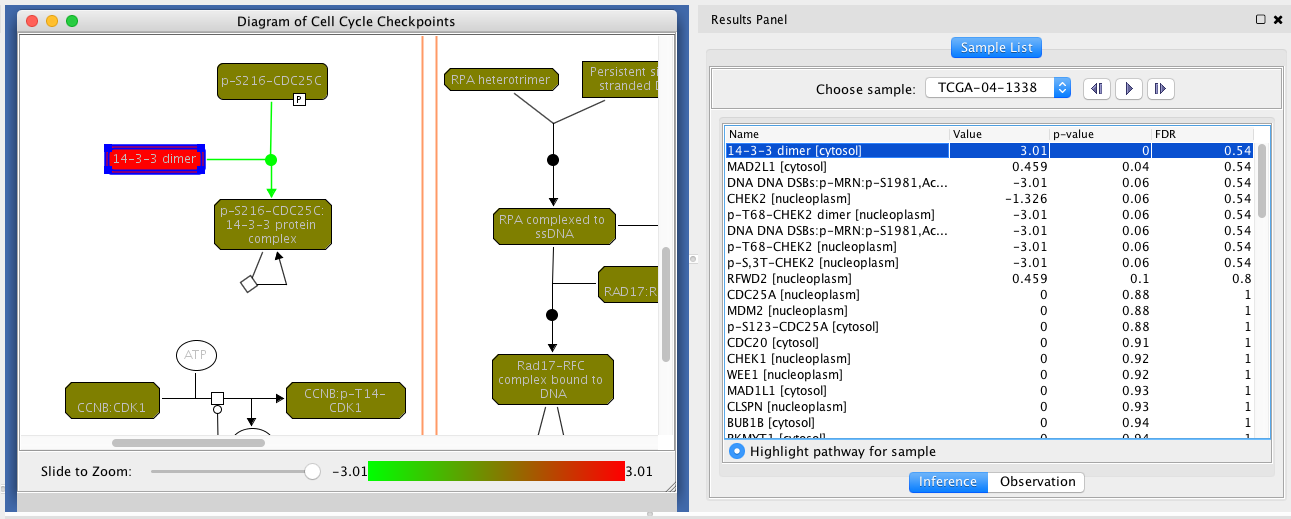 PGM示例视图:推论
PGM示例视图:推论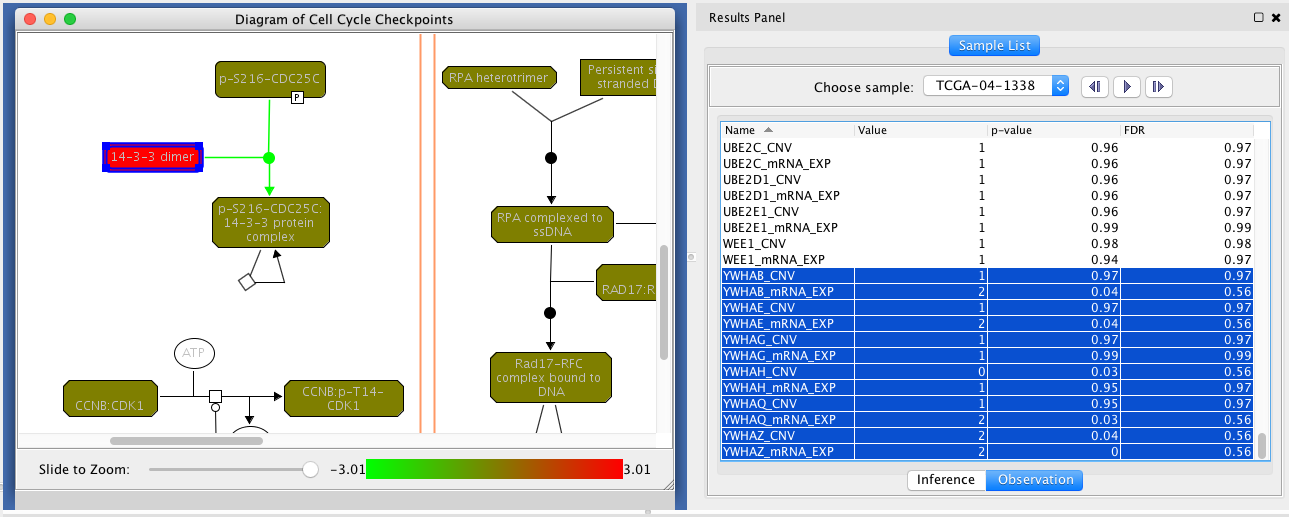 PGM示例视图:观察
PGM示例视图:观察 - 比较两个样本的分析结果:你可以比较两个样本的观察数据和推断结果。为此,在底部结果窗格的“IPA样本分析”选项卡中选择两个样本,然后使用弹出菜单“比较样本”,弹出另一个名为“样本比较”的选项卡。您可以查看推理和观察数据的比较结果。
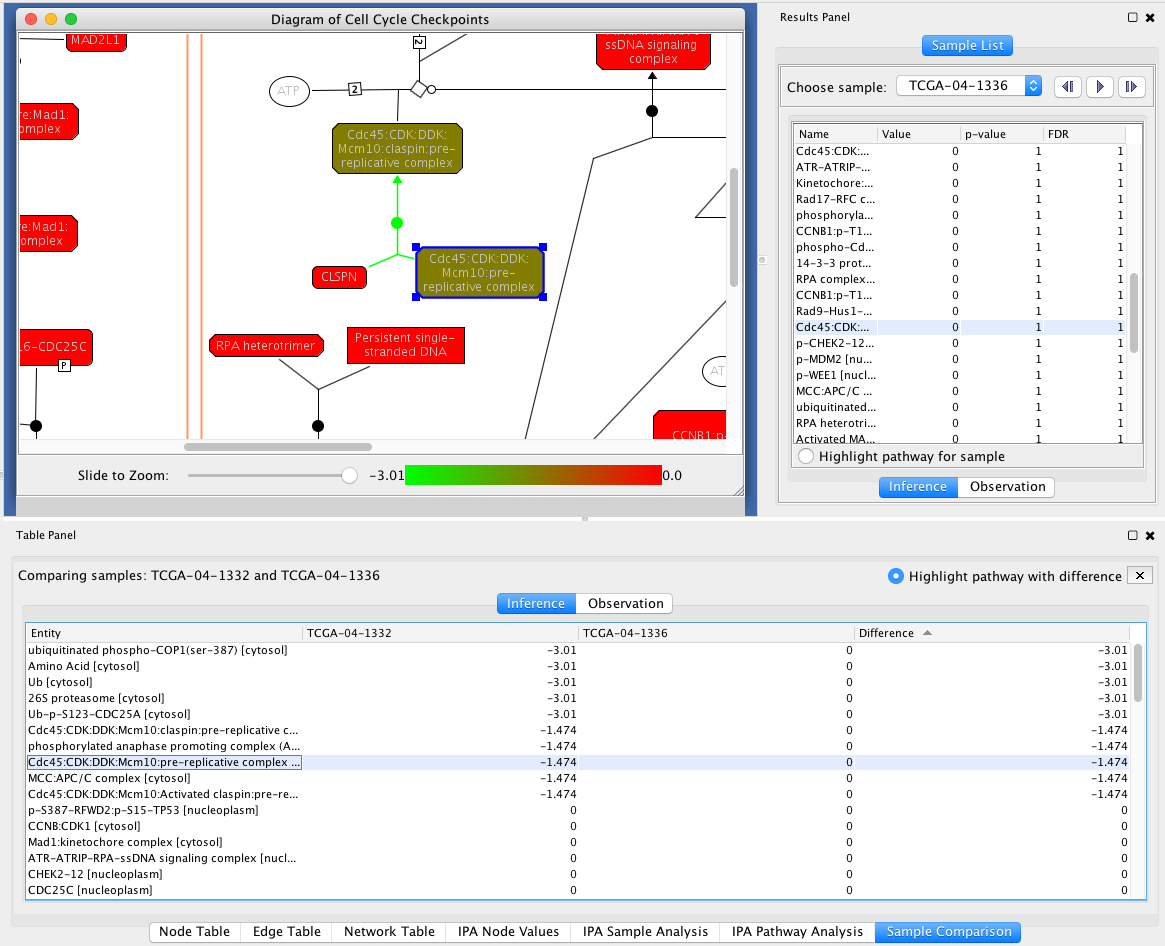 两样本比较
两样本比较











基于布尔网络的路径分析
我们开发了一种方法(Manuscript in preparation),将基于生化反应的Reactome路径转换为布尔网络,然后根据约束模糊逻辑方法进行路径模拟训练信号通路映射到生化数据与约束模糊逻辑:定量分析肝细胞对炎症刺激的反应和查询定量逻辑模型(Q2LM)研究细胞内信号网络和细胞因子相互作用.基于此方法,用户可以使用基于布尔网络构建的模糊逻辑的丰富反应体路径在Cytoscape内进行路径模拟。
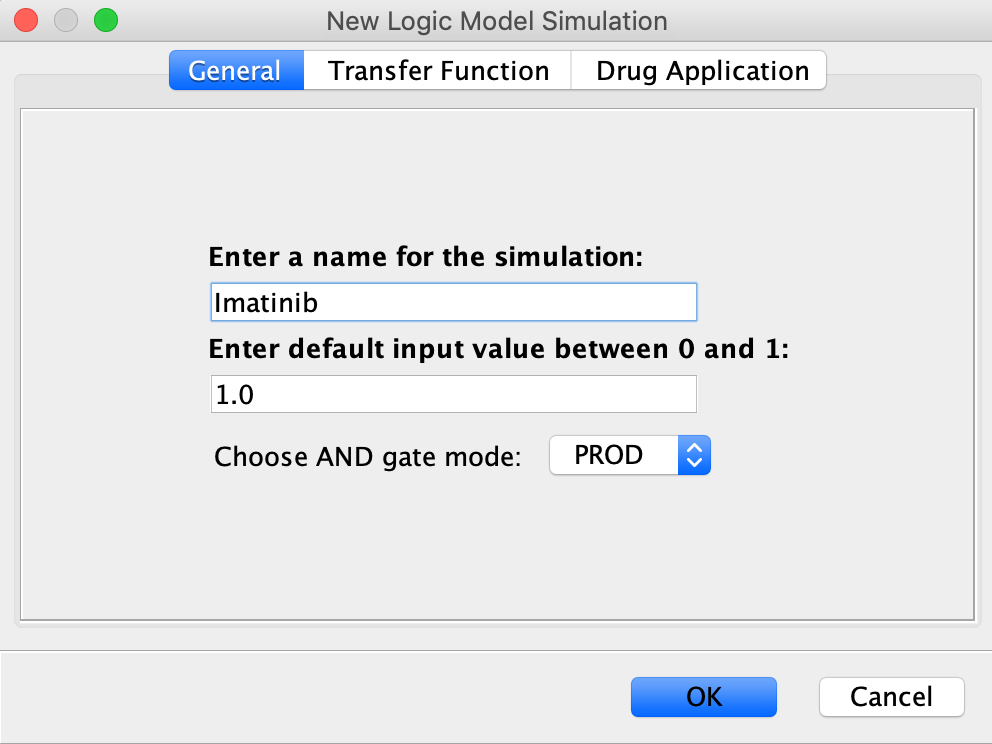
- 建立并运行逻辑模型仿真:在路径图视图中选择弹出菜单“运行逻辑模型分析”,得到新的模拟对话框。为模拟输入一个名称和默认值(通常应该是1.0以允许模拟继续进行),然后为and门模式选择PROD或MIN(默认选择PROD通常就可以了)。
请注意:您还可以选择传递函数并调整Hill功能的参数。但是,为简单起见,建议首先使用“身份函数”。申请如何应用逻辑模型仿真,见下文。
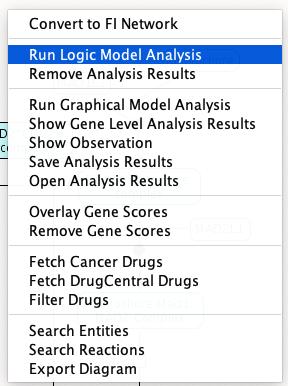 运行布尔网络分析
运行布尔网络分析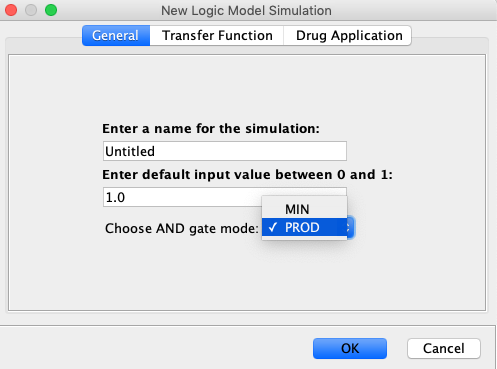 新BN模拟
新BN模拟
单击“新建模拟”对话框中的“确定”按钮后,默认初始配置将显示在“结果”面板中。通过单击所选变量的单元格,可以在设置表中更改变量类型和修改。要运行模拟,请单击“结果”面板中的“模拟”按钮。
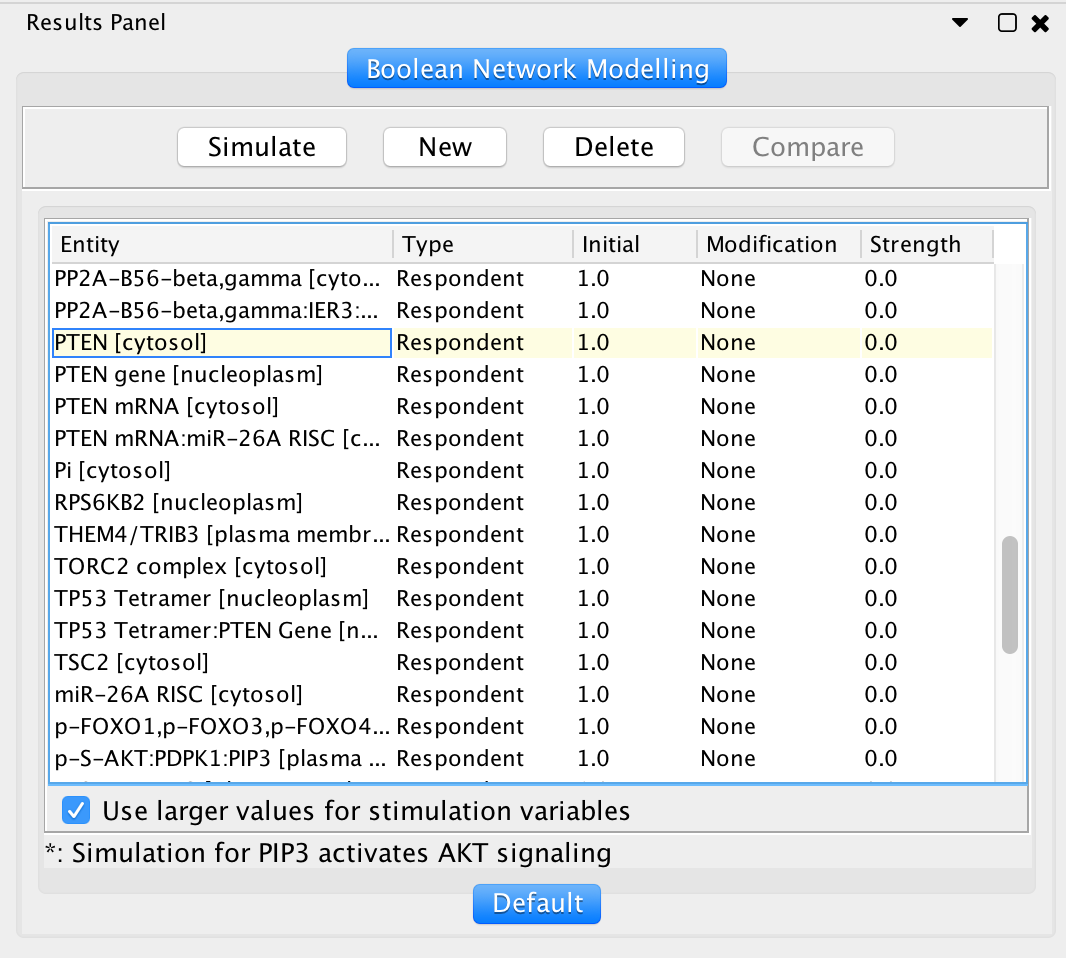 建立布尔网络分析
建立布尔网络分析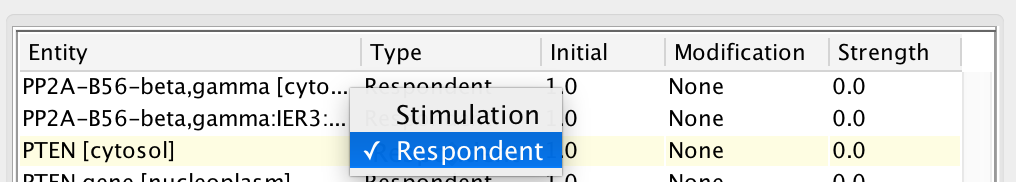 选择BN变量类型
选择BN变量类型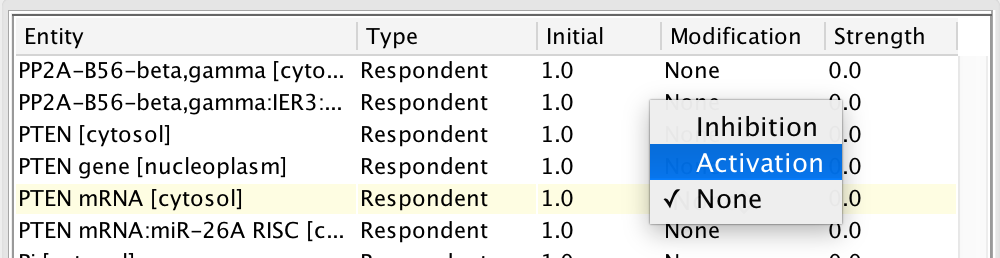 选择BN修改类型
选择BN修改类型 - 可视化模拟结果:模拟完成后,路径图中的实体将根据模拟值高亮显示,模拟值应介于0和1之间。您可以选择一个或多个实体,以在Cytoscape底部的表面板中可视化它们的时间行为。模拟计算的吸引子也列在结果面板内原始设置表的右列中。请注意:模拟后,您将无法修改任何初始配置。
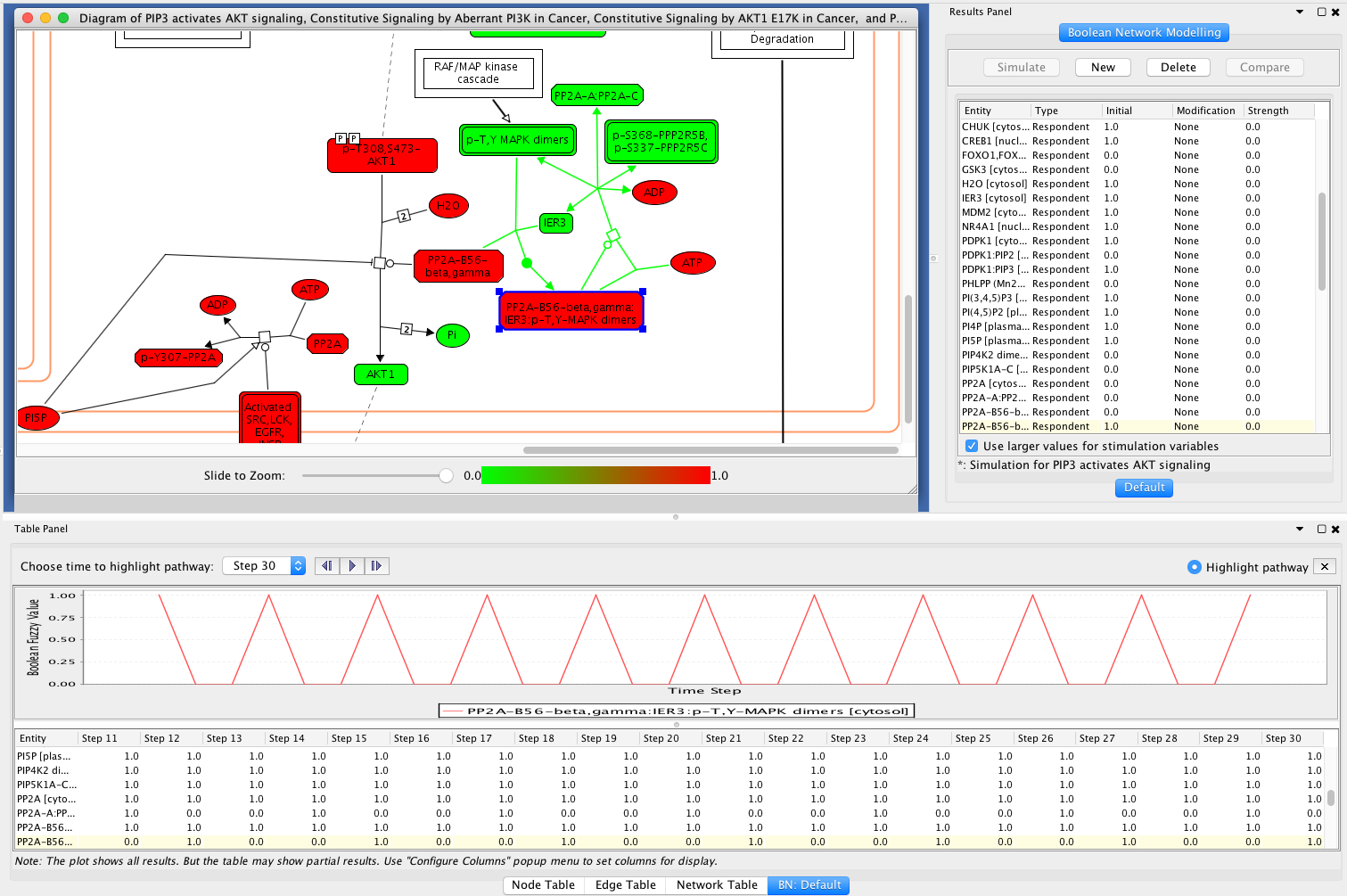 布尔网络仿真结果
布尔网络仿真结果
请注意:为了避免混乱,如果为一个逻辑模型模拟生成了太多的时间步长,则表中只显示最近的20个时间步长。然而,这个情节显示了所有的时间步骤。您可以选择不同的列显示在表中使用弹出菜单“配置列”后,在表中选择任何变量。 - 通过修改进行路径模拟:布尔网络模拟可以帮助用户发现实体活动的修改(例如,由体细胞突变引起的抑制或激活)对路径行为的影响。例如,在PIP3激活AKT信号中,复合物AKT:PIP3与EntitySet THEM4/TRIB3形成一个复合物,以形成另一个复合物,该复合物抑制AKT的激活(有关详细信息,请参阅Reactome PIP3激活AKT信号).要执行带有修改的模拟,请在模拟设置表中选择修改类型,并将强度分配给修改。
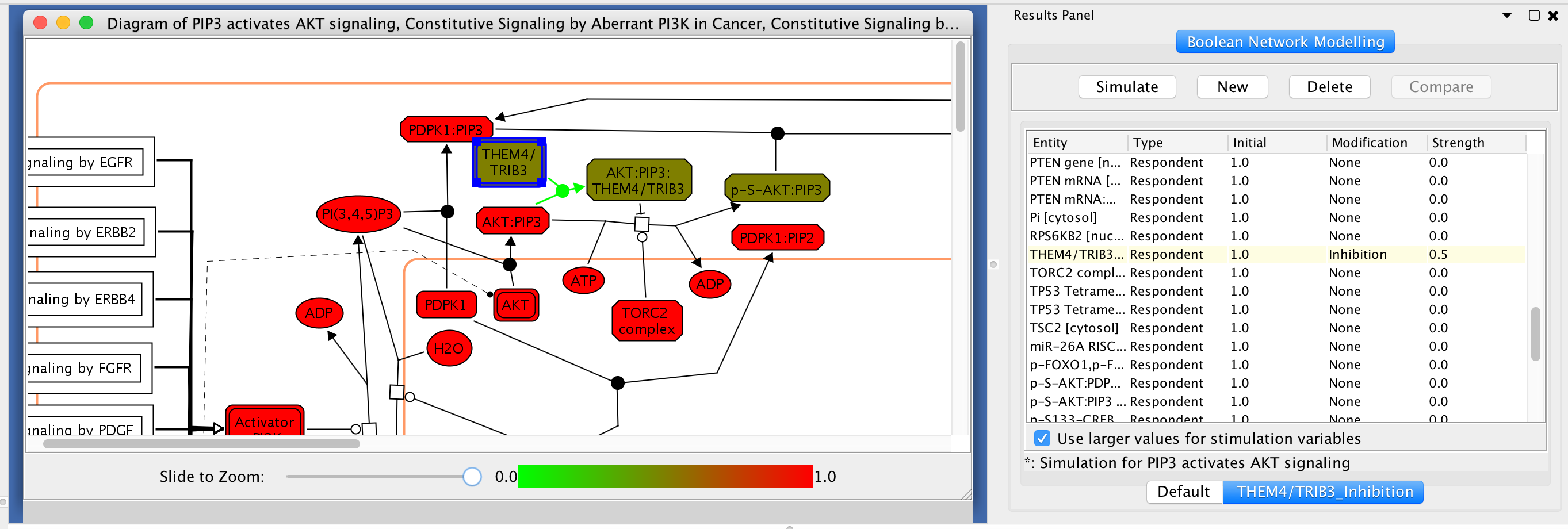 布尔网络仿真选择抑制
布尔网络仿真选择抑制
单击Simulate按钮,使用此配置的抑制调用受约束的模糊逻辑模型模拟。您可以通过选择一个实体,然后切换Cytoscape底部的模拟结果表,来比较两个配置之间的模拟结果。
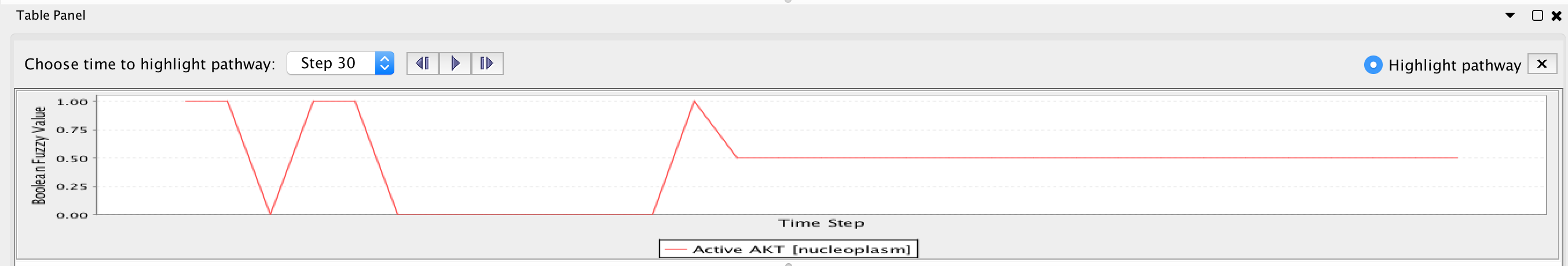 抑制AKT的活性
抑制AKT的活性
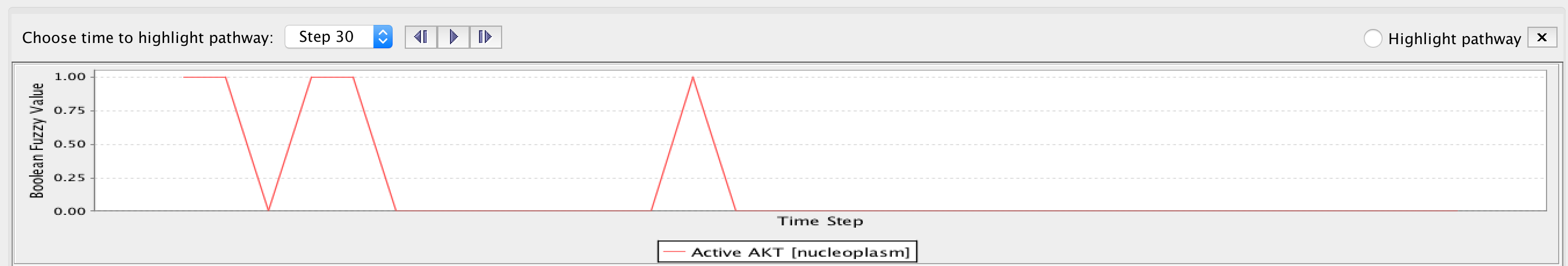 默认激活AKT
默认激活AKT
您还可以使用“结果”面板中的“比较”按钮获取比较对话框,然后选择两个模拟以进行比较。比较结果显示在底部表面板上列出的新表中。
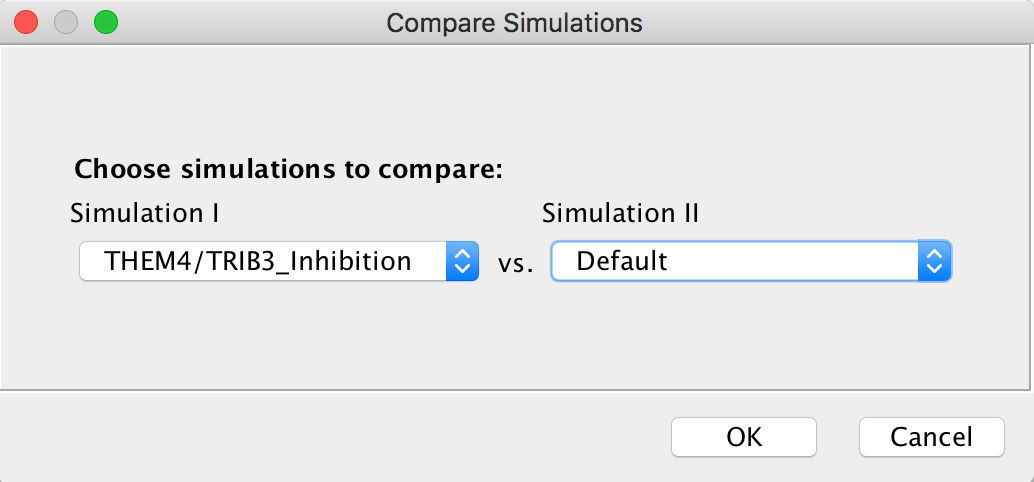 比较对话框
比较对话框
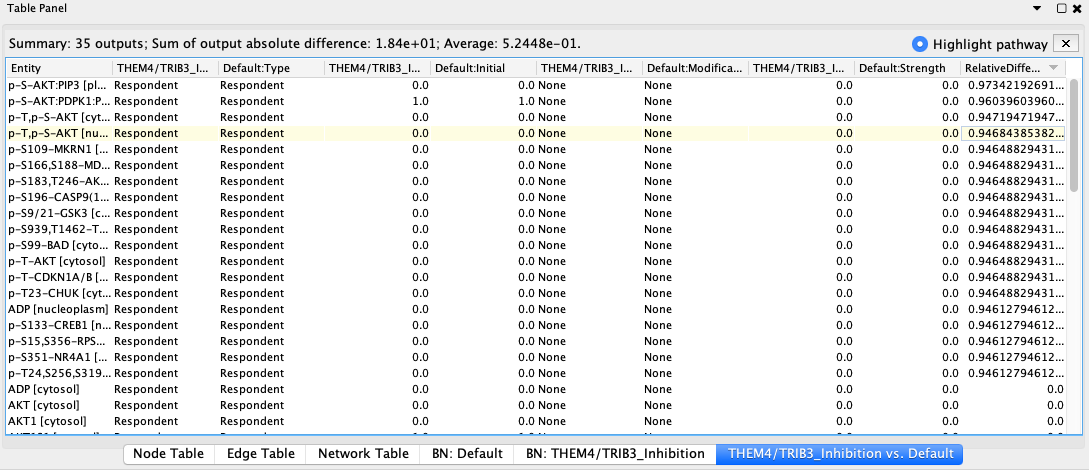 相比之下,活跃的akt
相比之下,活跃的akt
注意:比较结果表中的相对差值是根据每个模糊逻辑变量的相对变化计算的,计算为(valueInSim2 - valueInSim1) / (valueInSim2 + valueInSim1)。时间过程可以根据吸引子插值,直到相对的差异收敛为止。 - 要删除所有显示的受限模糊逻辑模拟结果,请使用弹出菜单“运行逻辑模型分析”下的“删除分析结果”。路径图应重新设置为原来的颜色,并删除所有与逻辑模型仿真相关的表。











反应体途径背景下结构变体的可视化
通过与博士合作。海德堡大学的Francesco Raimondi和Rob Russell利用蛋白质相互作用的三维结构提供Mechismo,是由罗素博士的小组为了研究单个氨基酸残基对蛋白质结构和功能的贡献,我们系统地分析了TCGA数据集中的突变,收集了一组反应和功能相互作用,这些反应和相互作用界面中显著富集突变的蛋白质(手稿准备中)。我们在ReactomeFIViz中添加了功能,以可视化这些三维结构和ReactomeFIViz路径、反应和相互作用中的突变残基。
- 在路径及其反应的背景下可视化分析结果:在打开的路径图视图中(见探索Reactome通路有关如何打开路径图),请使用弹出菜单“加载机械结果”将分析结果加载到打开的路径图中。加载结果后,根据底部表格中标记为“Mechanismo-Reaction”的FDR值对反应进行着色。请注意:您可以通过单击标记为“选择癌症类型的癌症类型”以突出基于表中的FDRS突出显示反应的突出显示癌症类型的底部选项卡顶部的列表来选择不同的癌症类型或康乃伊的结果。一些反应不受任何颜色突出显示,因为没有发现用于这些反应的蛋白质的结构变体。要从路径图中删除加载的结果,请使用另一个弹出菜单“删除机电源”。
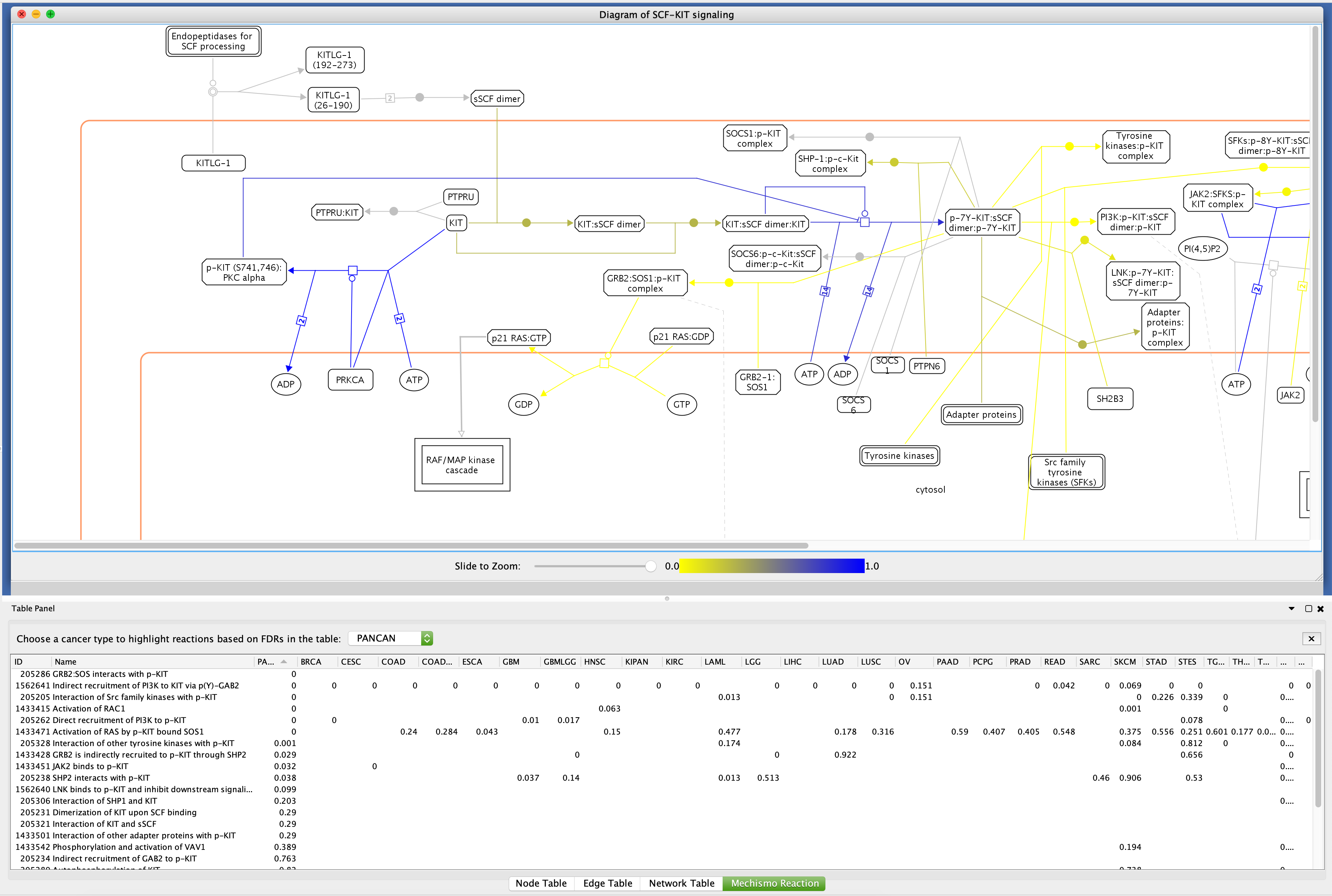 机械摩擦反应视图
机械摩擦反应视图 - 在通路FI网络的背景下可视化分析结果: Mechismo结果加载到路径图后,可以像往常一样通过弹出菜单“convert to FI network”将路径图转换为FI网络。FI网络视图中显示的边缘根据底部表中标有“Mechismo Interaction”的FDR值高亮显示。请注意:为了使FI网络视图更简单,在路径图视图的左下角勾选“Show FI Only for Selected”。您可以选择一个反应或复杂,然后在路径图视图中查看为所选对象提取的FIs。一些反应和复合物可能不包含任何FI(例如,蛋白质和化学物质组成的复合物或蛋白质和化学物质之间的反应)。
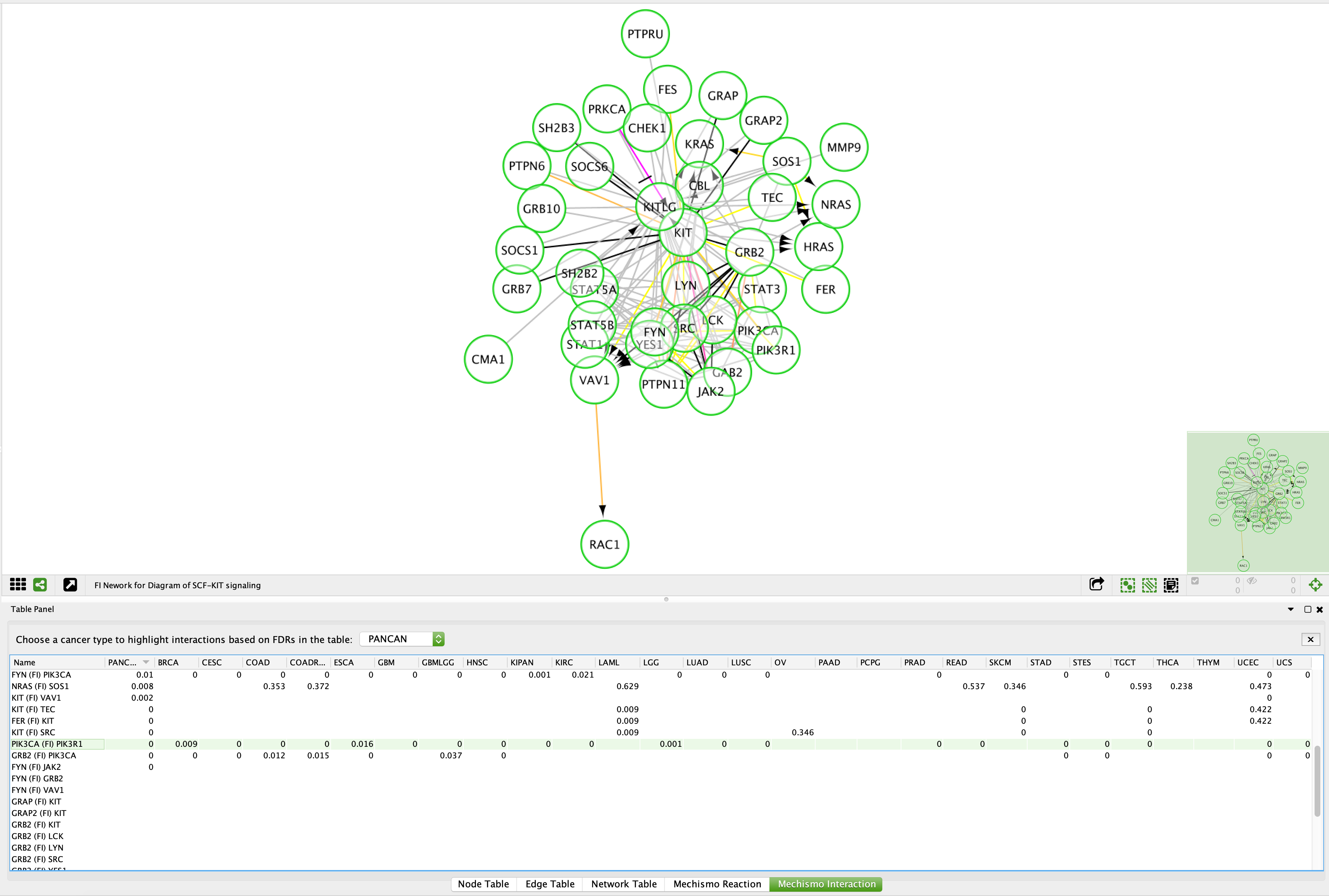 机械交互视图
机械交互视图 - 可视化蛋白质-蛋白质3D结构中的结构变体:在“网络视图”中,选择称为“获取机制结果”的弹出菜单,以使蛋白质蛋白质交互3D模型的视图。从TCGA数据集收集的结构变体映射到原始的蛋白质 - 蛋白质相互作用3D结构基于Mechismo平台。请注意:在结构视图中,将坐标映射到结构变体的氨基酸残基以球的形式显示。映射到底部表中选定行的留数用黄色突出显示。ReactomeFIViz使用Jmol用于蛋白质三维结构可视化。有关如何使用Jmol的更多信息,请参阅其文档:http://jmol.sourceforge.net/docs/.
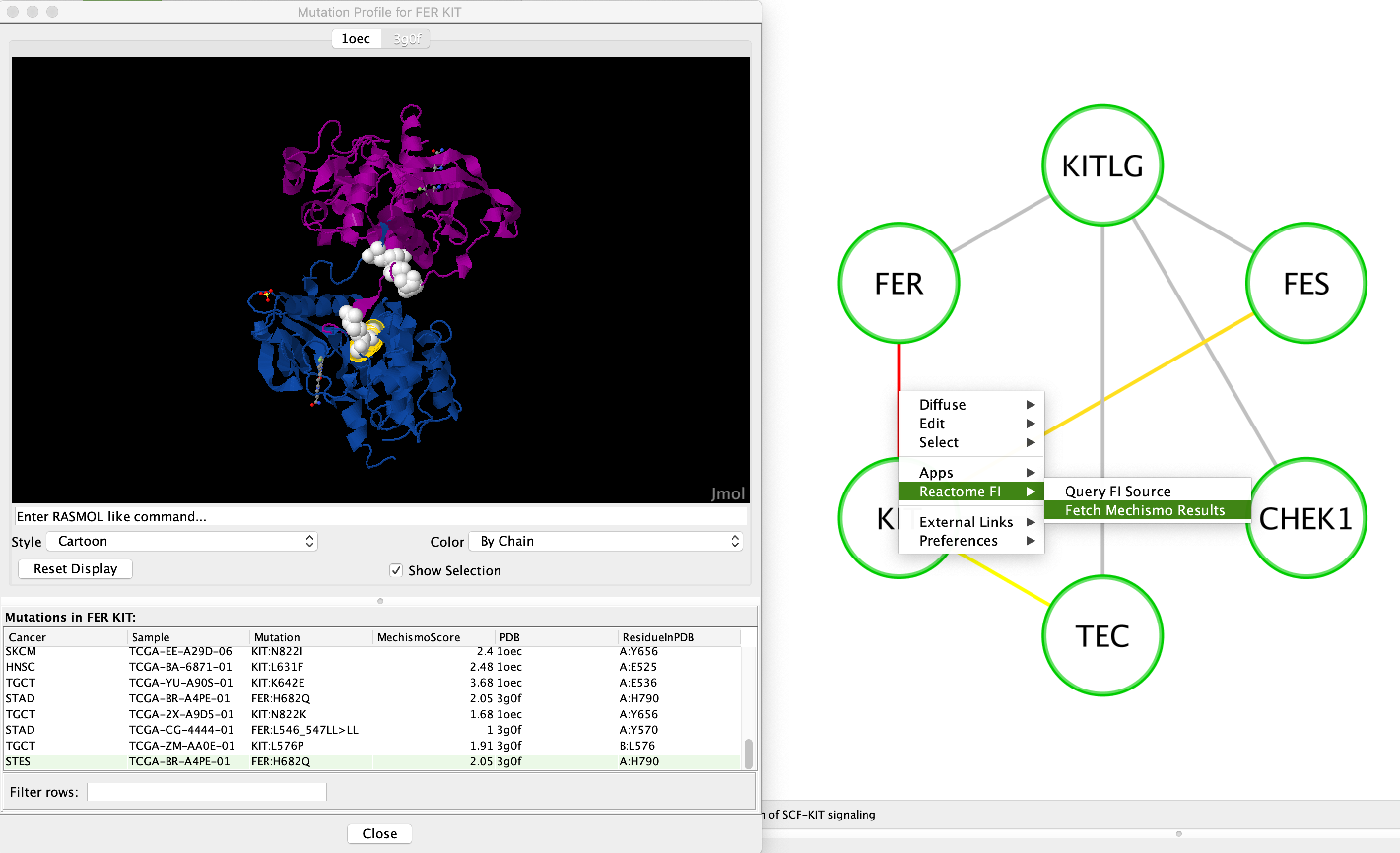 Mechismo结构视图
Mechismo结构视图



使用交互功能(FI)网络
安装ReactomeFIViz应用程序后,您应该会在应用程序菜单下看到一个名为“Reactome FI”的菜单项。单击此菜单,您将看到6个子菜单:基因设置/突变分析,的PGM影响分析,微阵列数据分析,反应途径和用户指南。基因集/突变分析用于对一组基因或突变数据文件进行基于网络的数据分析,PGM影响分析用于使用多个OMICS数据类型的反垃圾件FI网络的概率图形模型进行功能影响分析,热网算法搜索网络模块的热网突变分析(参见http://compbio.cs.brown.edu/projects/hotnet/),微阵列数据分析做MCL(马尔可夫图聚类,http://micans.org/mcl/)的FI网络聚类分析,将非加权FI网络转换为加权网络,利用网络中基因之间的相关性,Reactome通路从Reactome数据库加载通路,直接在Cytoscape中可视化Reactome通路,并进行通路富集分析。用户指南给你带来了这个用户指南。
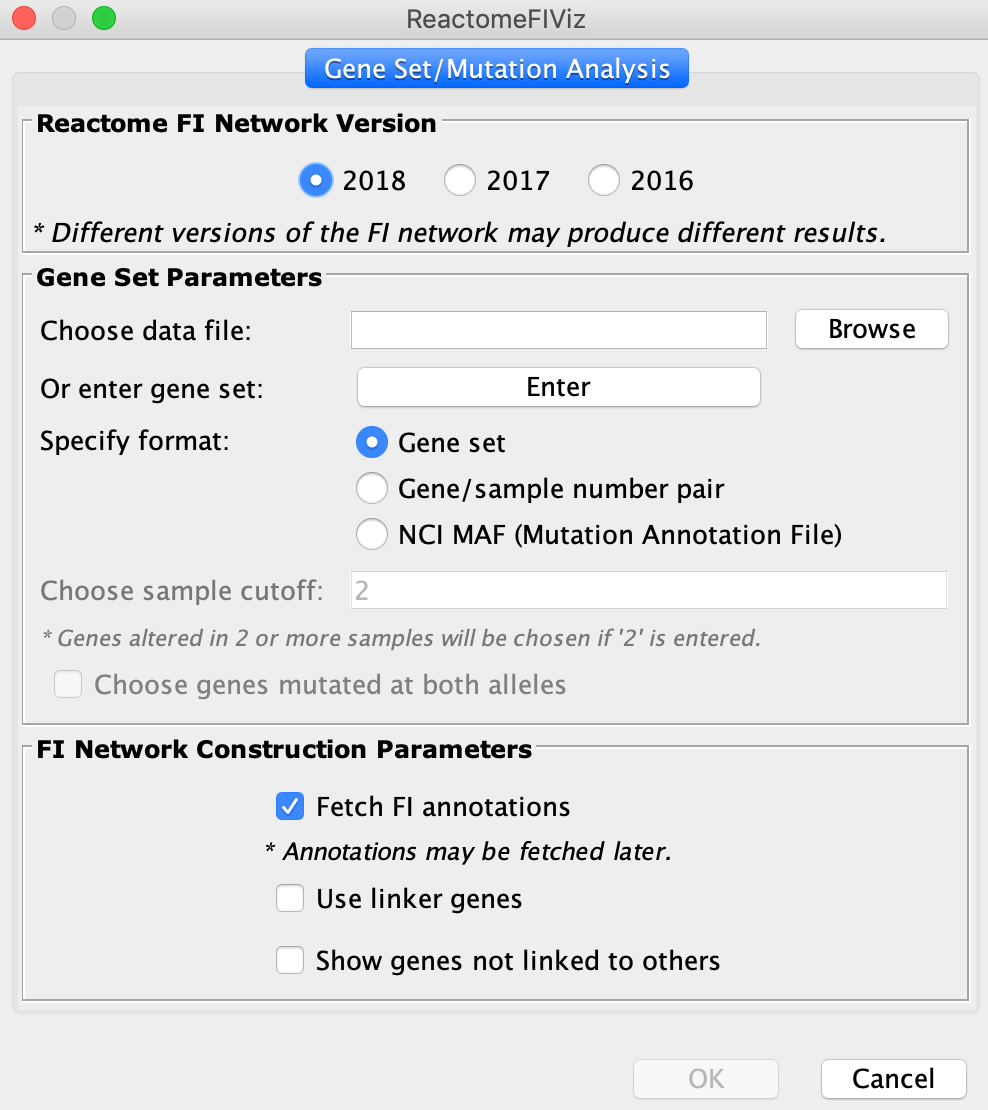
基因设置/突变分析
- 您可以通过单击“回车”按钮将基因列表直接输入ReactomeFIViz,或从本地文件加载。目前,ReactomeFIViz支持三种用于基因集/突变分析的文件格式:
- 简单的基因集:每个基因一株。例如,GWASFuzzyGenes.txt, T2D GWAS基因列表。
- 基因/样本数对.例如,GeneSampleNumber.txt,其中包含两个必填列,即基因和发生基因突变的样本数,以及一个可选的第三列,列出样本名称(以“;”分隔)。
- NCI MAF(突变注释文件).例如,GlioblastomaMutationTable.txt, TCGA GBM项目的突变文件。
- 从列出的三个版本中选择一个FI网络版本。
请注意:你可能会得到不同的结果使用不同的FI网络版本,因为一个较晚的版本可能包含更多的蛋白质/基因和更多的FI。但根据我们的经验,一个重要的FI网络模块在多个版本中通常是稳定的。 - 点击“确定”按钮直接输入基因,或者选择一个包含你想要用来构建功能交互网络的基因的文件。选择文件,选择合适的文件格式和参数,在对话框中加载基因,构建FI网络。点击“确定”按钮,开始FI网络建设过程。
- 构建好的FI网络将显示在网络视图面板中。将自动为FI网络创建一个FI特定的视觉风格。
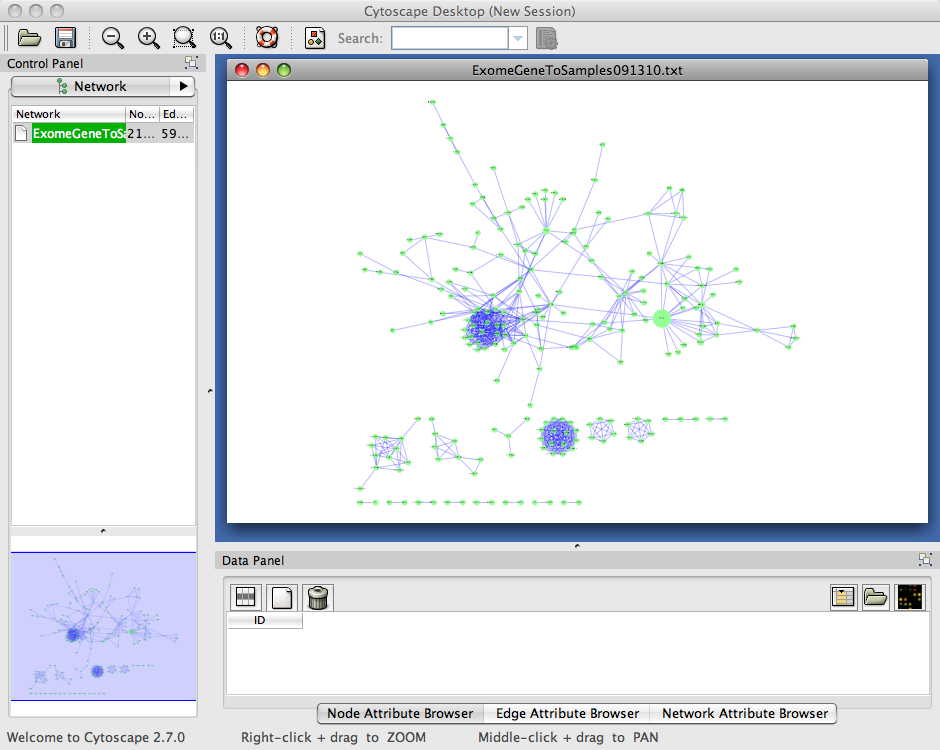 Reactome FI子
Reactome FI子 - 应通过弹出菜单调用Reactome FI插件的主要功能,右键单击网络视图面板中的空白区域即可显示弹出菜单。



- 获取fi注释:查询所选金融机构的详细信息。将创建三个与FI相关的边属性:FI注释、FI方向和FI分数。将基于FI方向属性值显示边。在下面的屏幕截图中,“->”表示激活/催化,“-|”表示抑制,“-”表示从配合物或输入物中提取的FIs,“-”表示预测的FIs。有关详细信息,请参见“VizMapper”选项卡、边源箭头形状和边目标箭头形状值。
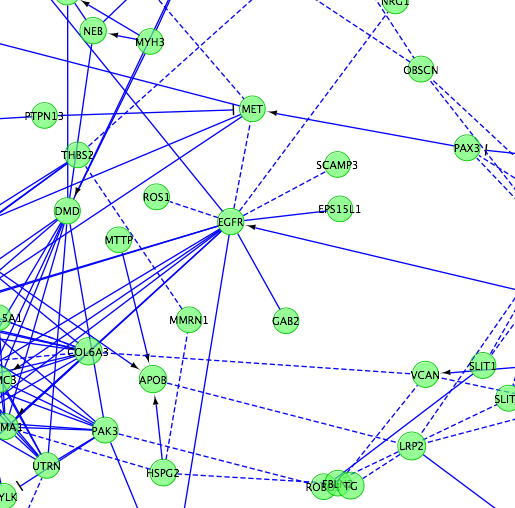 FI注释
FI注释
请注意:以下是有关显示列的简短说明:查询基因列表命中的Reactome FI网络中收集的路径的基因集;RATIOOFPROTENINESET:反应组FI网络中通路中包含的基因数与总基因数的比率;NumberOfProteinineset用于检测通路中的基因数量;用于查询基因列表中命中基因数的PROTEINFOROMNetwork;基于二项检验计算的P值;使用Benjamini-Hocherberg方法基于p值计算的FDR的FDR;通路中hit基因的节点。 - 分析网络功能:显示网络的路径或GO项富集分析。您可以选择通过FDR截止值来过滤浓缩结果。你也可以选择在网络面板中显示选定行或行中的节点,通过检查“隐藏未选定行中的节点”。每个途径基因集名称后括号中的字母对应路径注释的来源:C - CellMap, R - Reactome, K - KEGG, N - NCI PID, P - Panther, B - BioCarta。下面的截图显示了途径富集分析的结果。
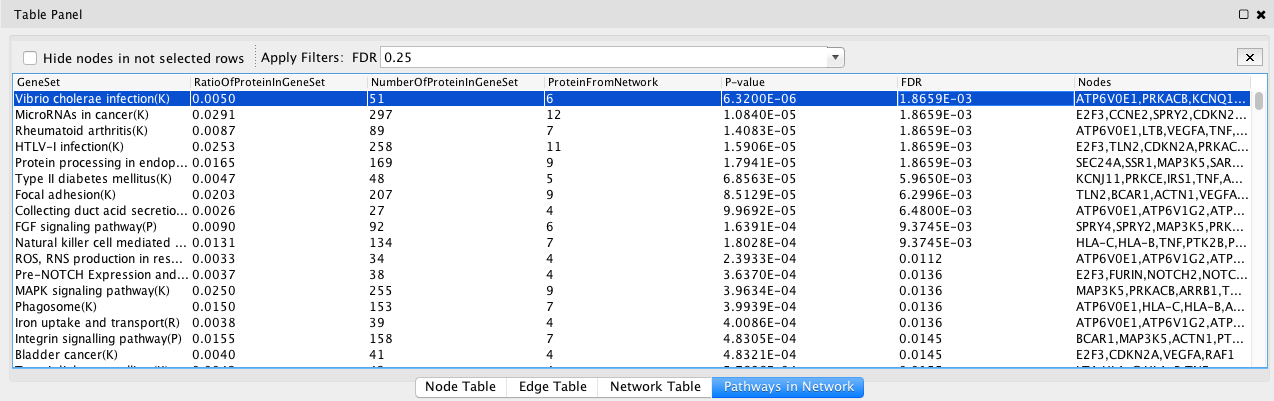 FI子网络中的通路
FI子网络中的通路
提示:要分析一组没有连接在一起的基因上的通路或GO term富集,请在“设置FI网络参数”对话框中选择“显示与他人没有连接的基因”选项。 - 集群FI网络:运行网络聚类算法(基于谱划分的网络聚类由纽曼2006)在显示的FI网络上。不同网络模块中的节点将以不同的颜色显示(根据大小,前15个模块仅使用不同的颜色)。
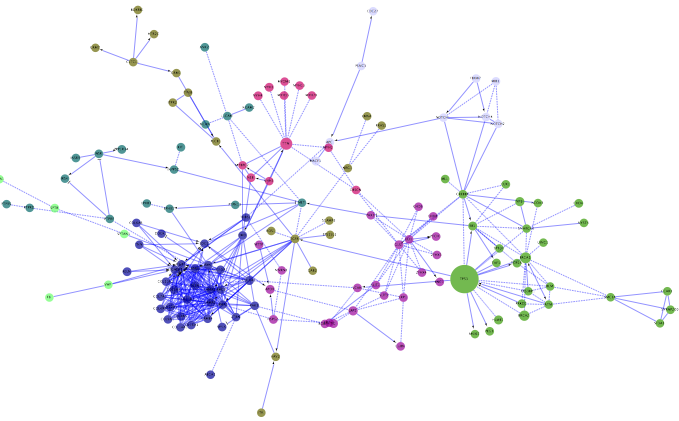 网络模块
网络模块 - 分析模块功能:每个单独网络模块的通路或GO项富集分析。您可以选择一个大小截止点来过滤掉过小的网络模块,选择一个FDR截止点来查看特定FDR值下的富集路径或GO项,并在选定的行中查看节点,或者只查看网络图中的行。
- 分析一组选定基因的功能:选择网络视图中显示的一组节点,然后选择弹出菜单“Analyze nodes Functions”,进行路径或GO term富集分析。结果将显示在一个对话框中。
- 载荷癌基因指数:负载癌症基因索引注释。详细信息请参见章节载荷癌基因指数.



的PGM影响分析
- 我们开发了一种基于概率图形模型(PGM)的功能影响分析,利用Reactome FI网络将多种组学数据类型集成在一起。当前版本的ReactomeFIViz支持四种组学数据类型:CNV、mRNA表达、DNA甲基化和体细胞突变。用于此分析的铂质谱是基于马尔可夫随机场(MRF).目前我们支持两种类型的MRFS:成对MRF和最近的邻居吉布斯MRF. 您可以从这两种型号中选择一种。我们推荐成对MRF,因为它简单。
- 要执行基于PGM的功能影响分析,请选择菜单,Apps/Rectome FI/PGM影响分析/Analyze。您可以使用PGM配置对话框输入您的组学数据。
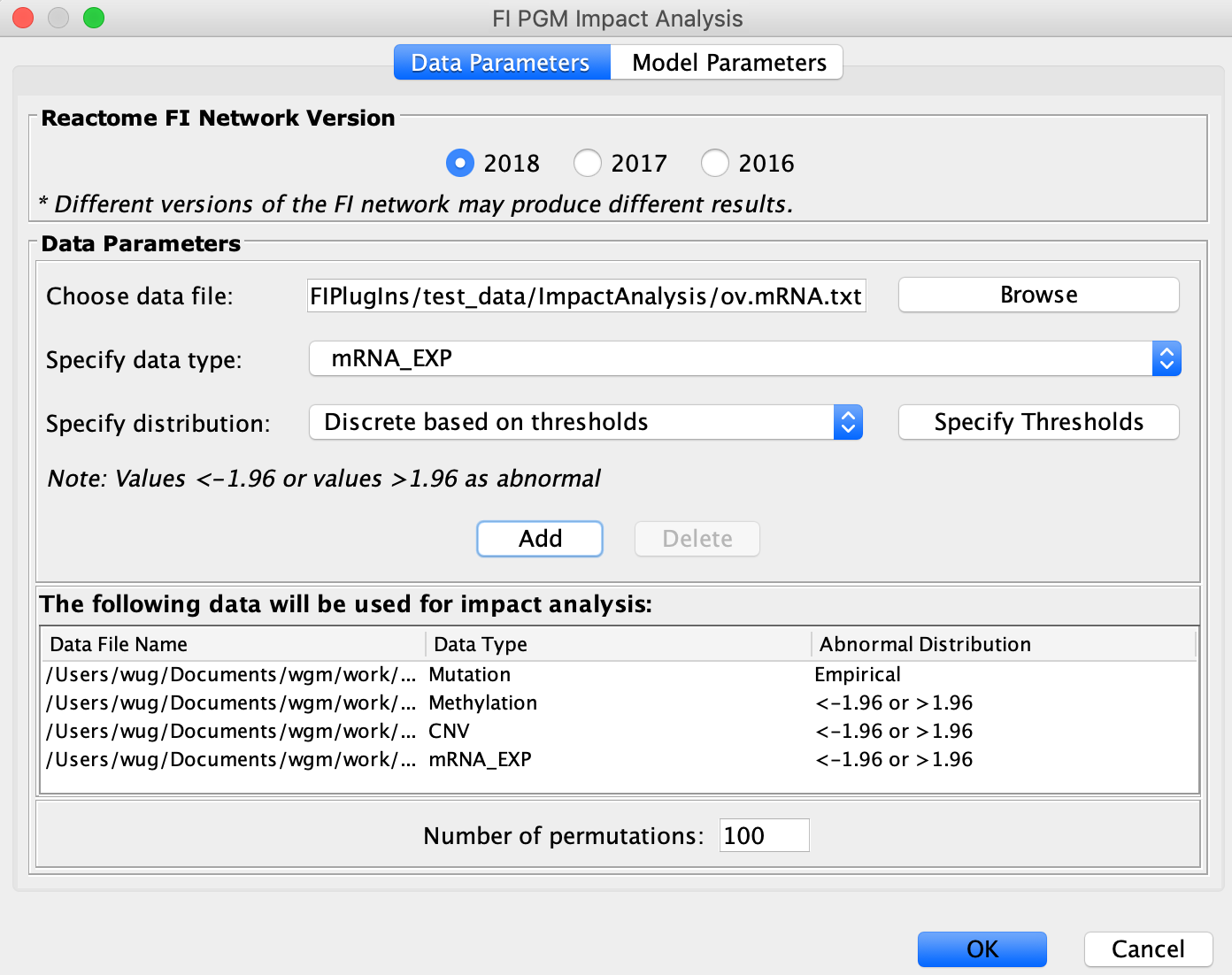 FI-PGM配置对话框
FI-PGM配置对话框
笔记:
1). ReactomeFIViz支持连续观测变量,不需要离散化,在配置中选择“使用经验分布”。对于体细胞突变,目前它只支持NCI MAF文件格式,并需要一个名为“MA_FI”的特定列。“得分”为突变函数的影响得分突变评估员或者从其他来源。
2).使用的默认MRF模型是PairwiseMRF。您可以为第一次测试选择默认设置。
3). MRF模型需要几个参数。我们根据一个小玩具模型调整了这些参数。在当前版本的ReactomeFIViz中,不能更改这些参数。 - 完成整个分析可能需要几个小时。实际运行时间取决于数据的大小。在下面的进度窗格中显示正在运行的作业的进度。你可以随时取消跑步。
 FI PGM运行进度
FI PGM运行进度 - 分析完成后,您应该会看到类似于下面截图的结果对话框。您可以使用过滤特征来筛选您想要用于构建FI子网络以进行进一步分析的基因列表。
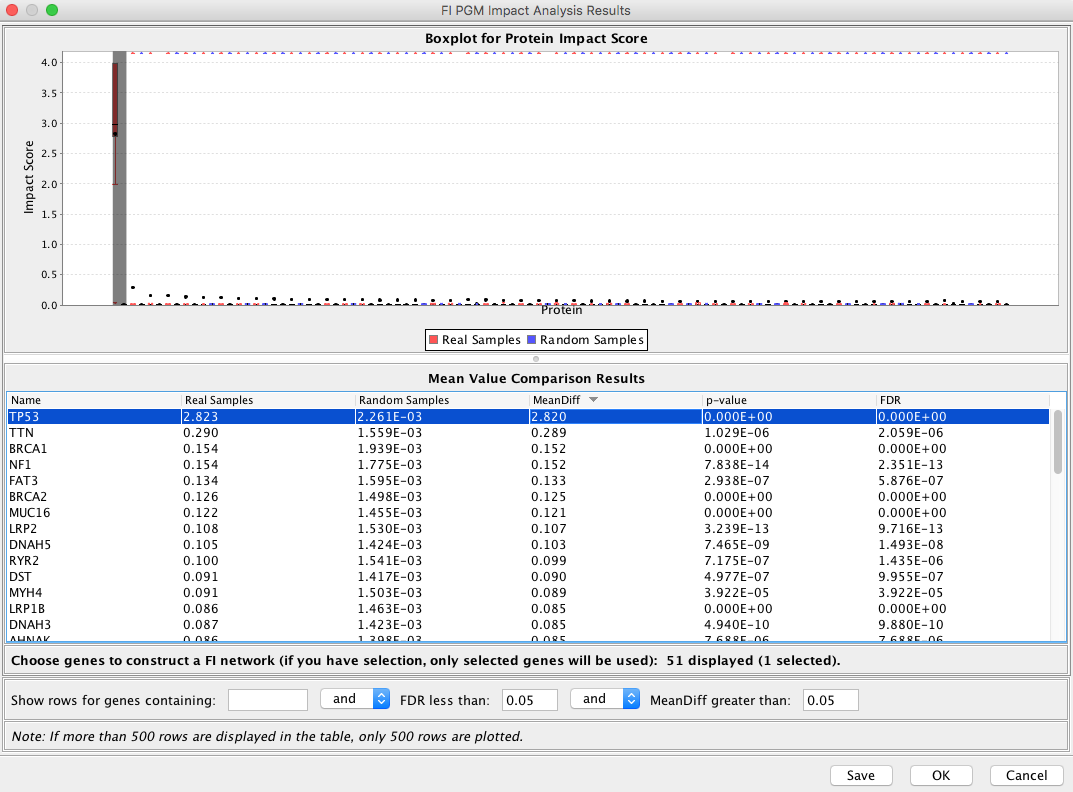 FI-PGM结果对话框
FI-PGM结果对话框
请注意:如果在结果表中选择一个或多个基因,则只使用这些被选择的基因来构建FI子网络。您可以通过点击“保存”按钮保存完整的分析结果(将保存所有基因的结果,而不仅仅是显示的结果)。我们强烈建议在点击“确定”按钮之前先保存您的结果,以便您可以访问您的结果。 - 选择“OK”按钮后,将构建一个FI子网,并显示在网络视图中。显示节点的大小与FI-PGM模型推导出的影响分数成正比。要查看影响分数和加载的观察数据,您可以在结果面板的“样本列表”选项卡中选择一个样本。您还可以通过检查sample List选项卡中的“为示例突出显示网络”来启用基于示例的网络可视化。如果您想查看用于构建FI子网络的原始结果,请单击“表格面板”中的“影响基因值”选项卡中的“显示所有结果”。
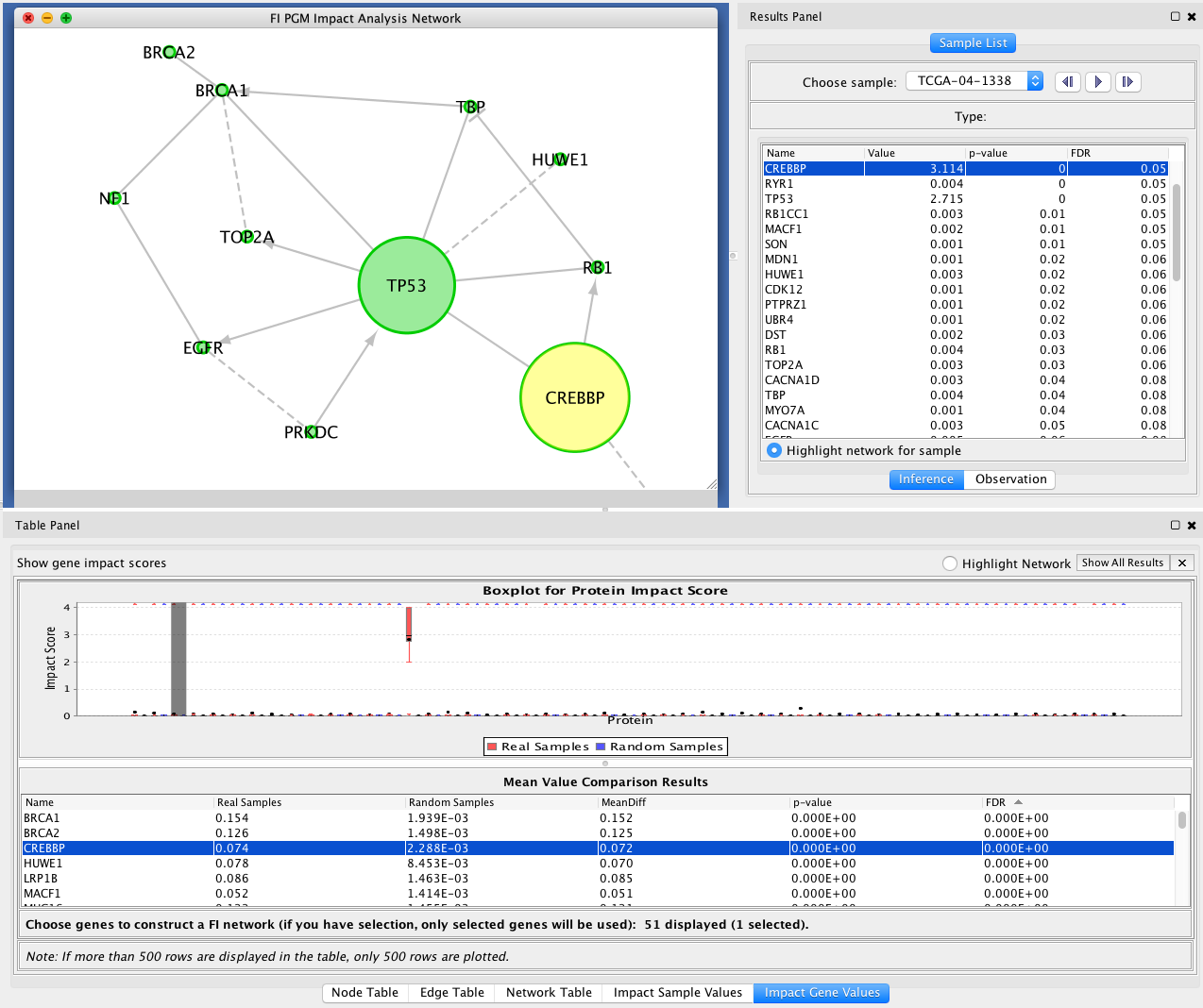 FI-PGM结果网络
FI-PGM结果网络




微阵列数据分析
ReactomeFIViz应用程序可以加载基因表达数据文件,计算同一FIs中涉及的基因之间的相关性,将计算出的相关性用作整个FI网络中边缘(即FIs)的权重,将MCL图聚类算法应用于加权FI网络,以及基于模块大小和平均相关性为所选网络模块的列表生成子网络。生成的FI子网络将显示在网络面板中,并可用于基因集/突变分析中的分析。有关此方法的详细信息,请参阅我们的出版物:基于网络模块的识别癌症预后特征的方法.
数组数据文件应该是一个带有表头的制表符分隔的文本文件。第一列应该是基因名。所有其他列应该是不同示例中的表达式值。文件中的数据集应预先规范化。例如,请看乳腺癌的基因表达文件:NejmLogRatioNormGlobalZScore_070111.txt.zip. 此数据集是从下载的van de Vijver等人的研究,并已标准化。
- 选择一个微阵列数据文件,运行MCL网络集群:在菜单Plugins/Reactome FIs中选择子菜单“Microarray Data Analysis”后,你应该会看到下面的对话框。选择一个微阵列数据文件,检查是否要使用绝对值作为边的权重,并为MCL聚类算法输入膨胀参数(-I)。膨胀参数越小,生成的网络模块的平均大小越大。根据我们自己的经验,我们用5.0作为膨胀参数,推荐值最高,边权值选择绝对值。有关如何选择膨胀参数的更多细节,请参阅http://micans.org/mcl/.设置好这些参数后,单击OK按钮,加载数据文件,计算相关性,并应用MCL聚类算法。
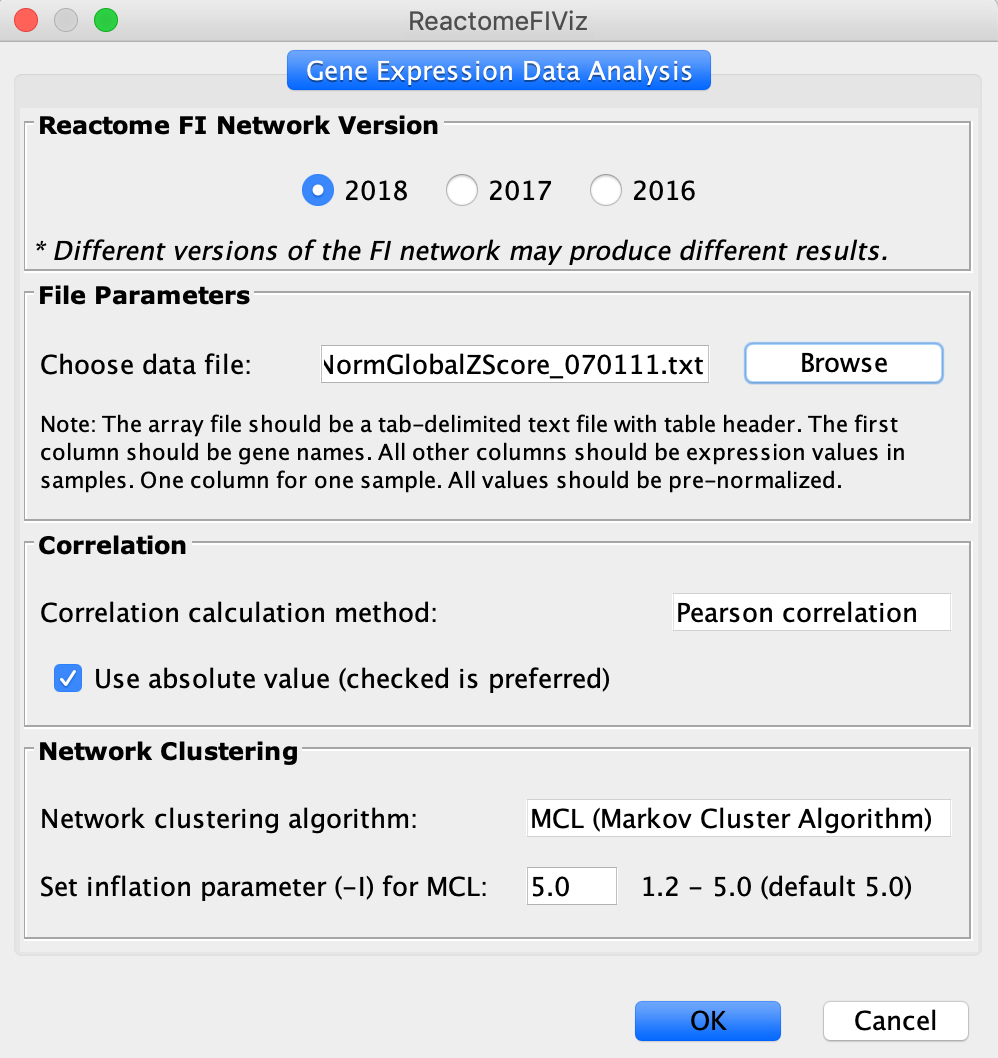 设置阵列数据分析参数
设置阵列数据分析参数 - 选择网络模块,建立FI子网生成的网络模块在MCL聚类结果对话框中列出(见下面)。只能列出2个以上基因的模块,用于FI子网络建设。您可以选择模块大小或平均相关值(绝对值,如果绝对值之前已经检查过),以过滤掉可能不重要的模块(注:设置这些截止值后,请按“Enter”键提交您的更改。)在我们的分析中,我们选择具有7个或更多基因且平均相关值不低于0.25的模块。这些值在对话框中被用作默认值。在对话框中,你可以看到有多少模块和基因将被选择在你选择的过滤值下建立FI子网络。单击OK按钮开始构建子网络。构建的子网络将被显示出来,并可以像基因集/突变分析生成的子网络一样进行分析。
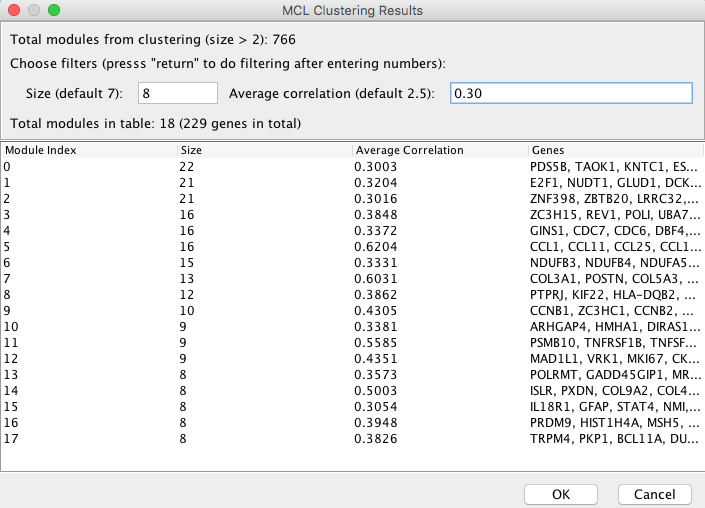 选择MCL模块
选择MCL模块


在反应体通路和FI网络中观察药物
ReactomeFIViz提供了一套功能,以帮助用户在reactomepathway和网络的背景下可视化药物。药物数据来源包括两个:Cancer Targetome (Blucher等人2017),其收集所有FDA批准的癌症药物(2018年之前)及其来自四种来源的目标相互作用,包括药物银行,治疗目标数据库,Iuphar和BindingdB;DrugCentral,一个全面的药物数据库支持NIH IDG程序.
在反应途径中可视化药物
- 列出所有FDA批准的抗癌药物:在Reactome路径树中使用弹出菜单“查看癌症药物”,在表格对话框中获得Cancer Targetome中收集的所有FDA批准的癌症药物列表。
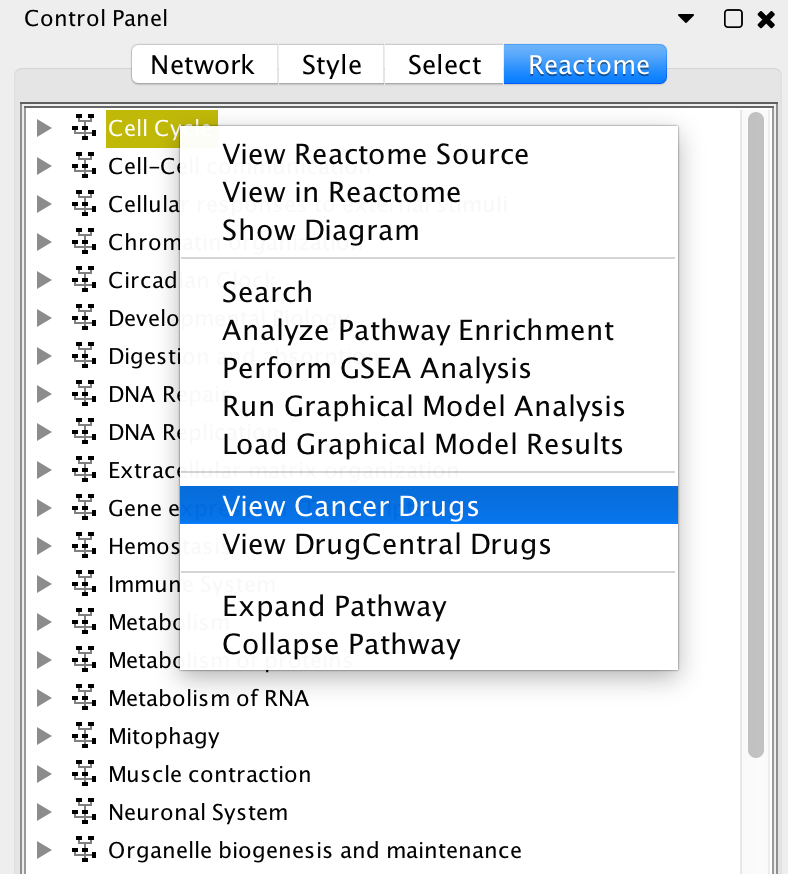 视图抗癌药物
视图抗癌药物
此药物列表的屏幕截图如下所示:
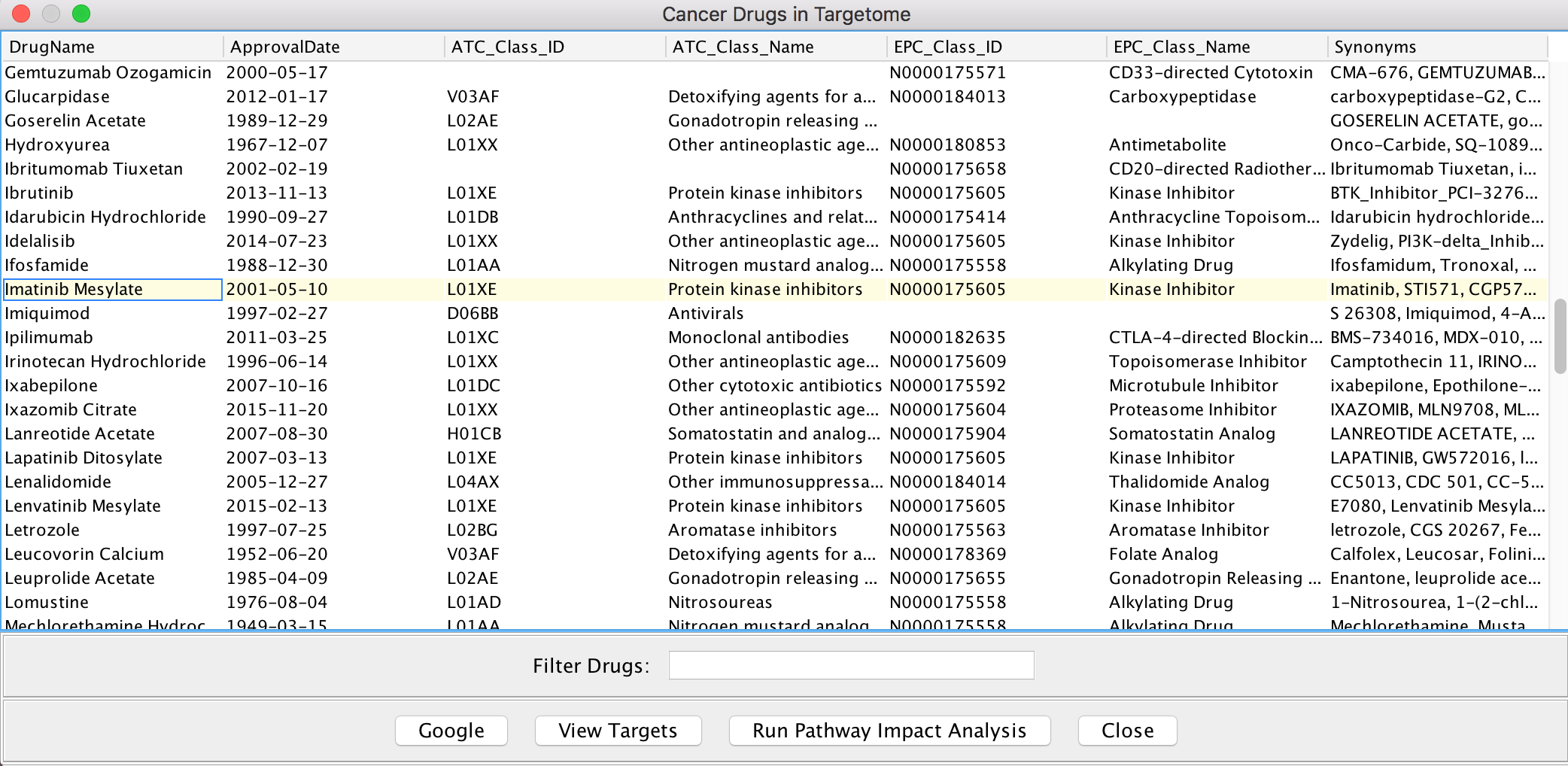 抗癌药物一览表
抗癌药物一览表 - 查看药物/靶点相互作用:在药物表中选择一种药物(见上图)以执行Google搜索,或通过单击对话框中的一个按钮来查看其目标。提供两个视图用于药物/目标相互作用。药物靶标表视图(下图)显示了所选药物的靶标具有收集的结合亲和力,其分为不同的类别(例如):KD,IC50,Ki和EC50。使用一组默认过滤器预先过滤显示的目标。您可以通过单击“过滤器”按钮更改“药物/目标交互过滤器”对话框中的默认过滤器。
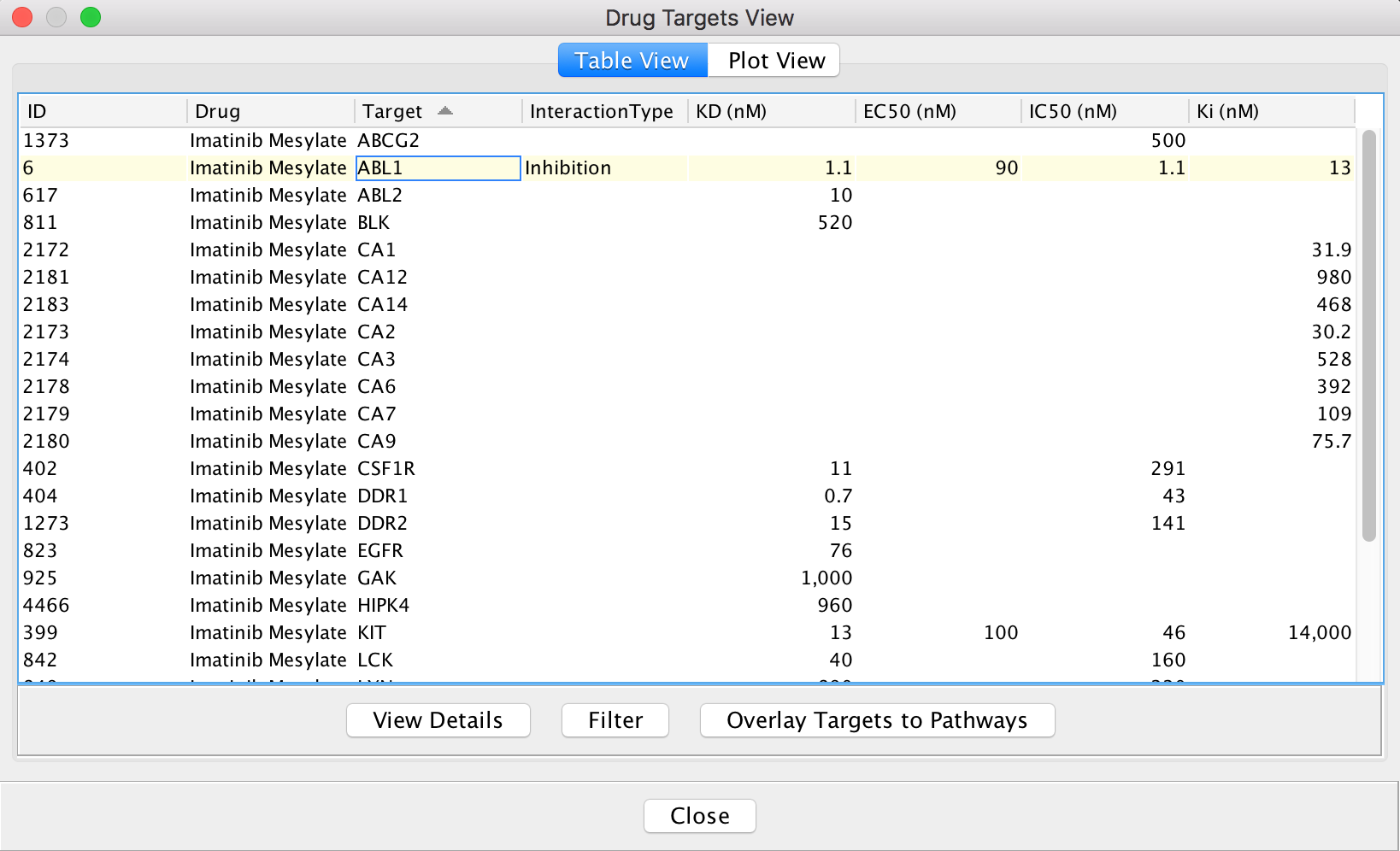 药物靶标表视图
药物靶标表视图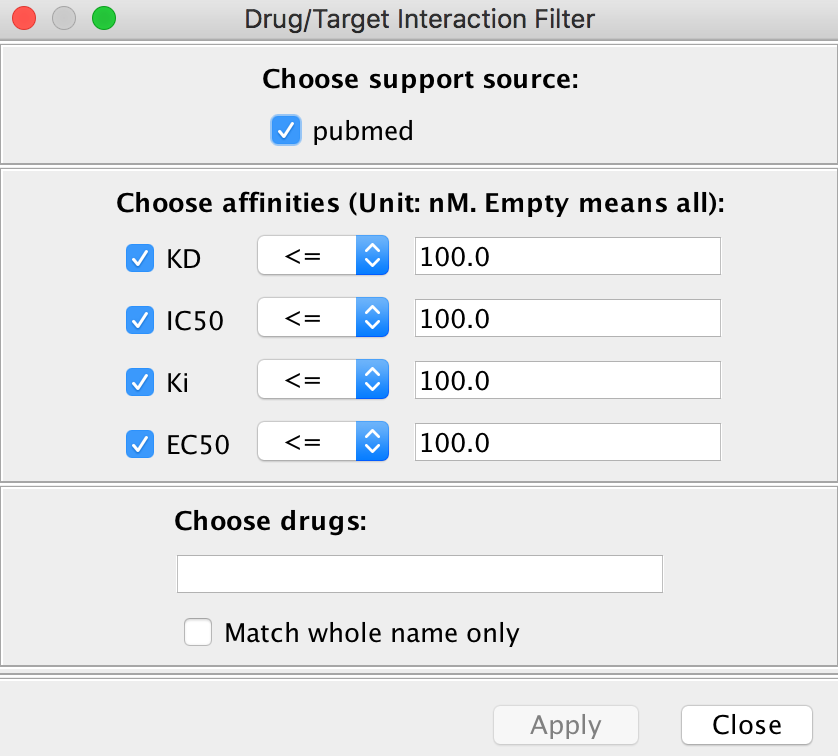 药物靶相互作用过滤器在“药物目标”表视图中,您可以选择交互,然后单击“查看详细信息”按钮以查看所选交互的详细信息。在“详细信息”视图中,您可以通过点击超链接浏览对实验证据的原始PubMed参考,或通过点击超链接来进行目标。
药物靶相互作用过滤器在“药物目标”表视图中,您可以选择交互,然后单击“查看详细信息”按钮以查看所选交互的详细信息。在“详细信息”视图中,您可以通过点击超链接浏览对实验证据的原始PubMed参考,或通过点击超链接来进行目标。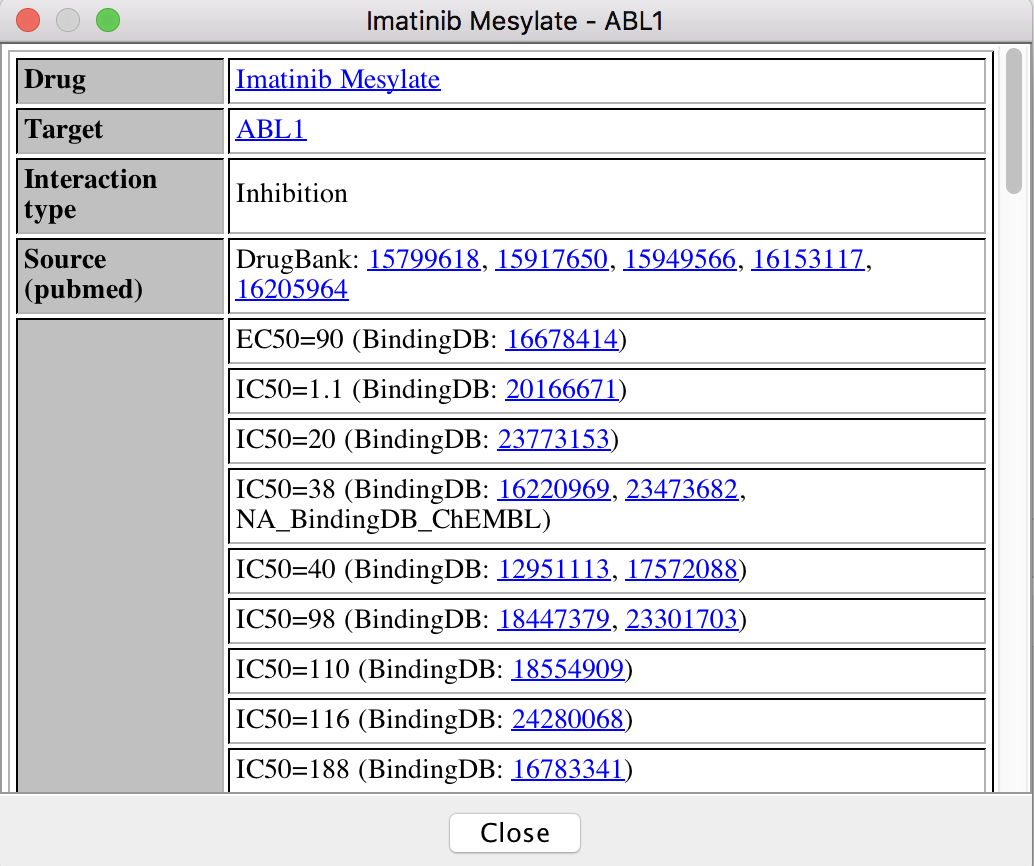 癌症药物相互作用详细信息视图你也可以选择表格中的多行,通过点击“覆盖目标到路径”来进行路径富集分析。看到通路富集分析路径富集分析结果详情。药物靶标图视图(下图)提供了一个分段的柱状图,显示了所选药物与其所有靶标之间的证据支持的相互作用,在一个图形视图中显示了主要靶标和次要靶标。
癌症药物相互作用详细信息视图你也可以选择表格中的多行,通过点击“覆盖目标到路径”来进行路径富集分析。看到通路富集分析路径富集分析结果详情。药物靶标图视图(下图)提供了一个分段的柱状图,显示了所选药物与其所有靶标之间的证据支持的相互作用,在一个图形视图中显示了主要靶标和次要靶标。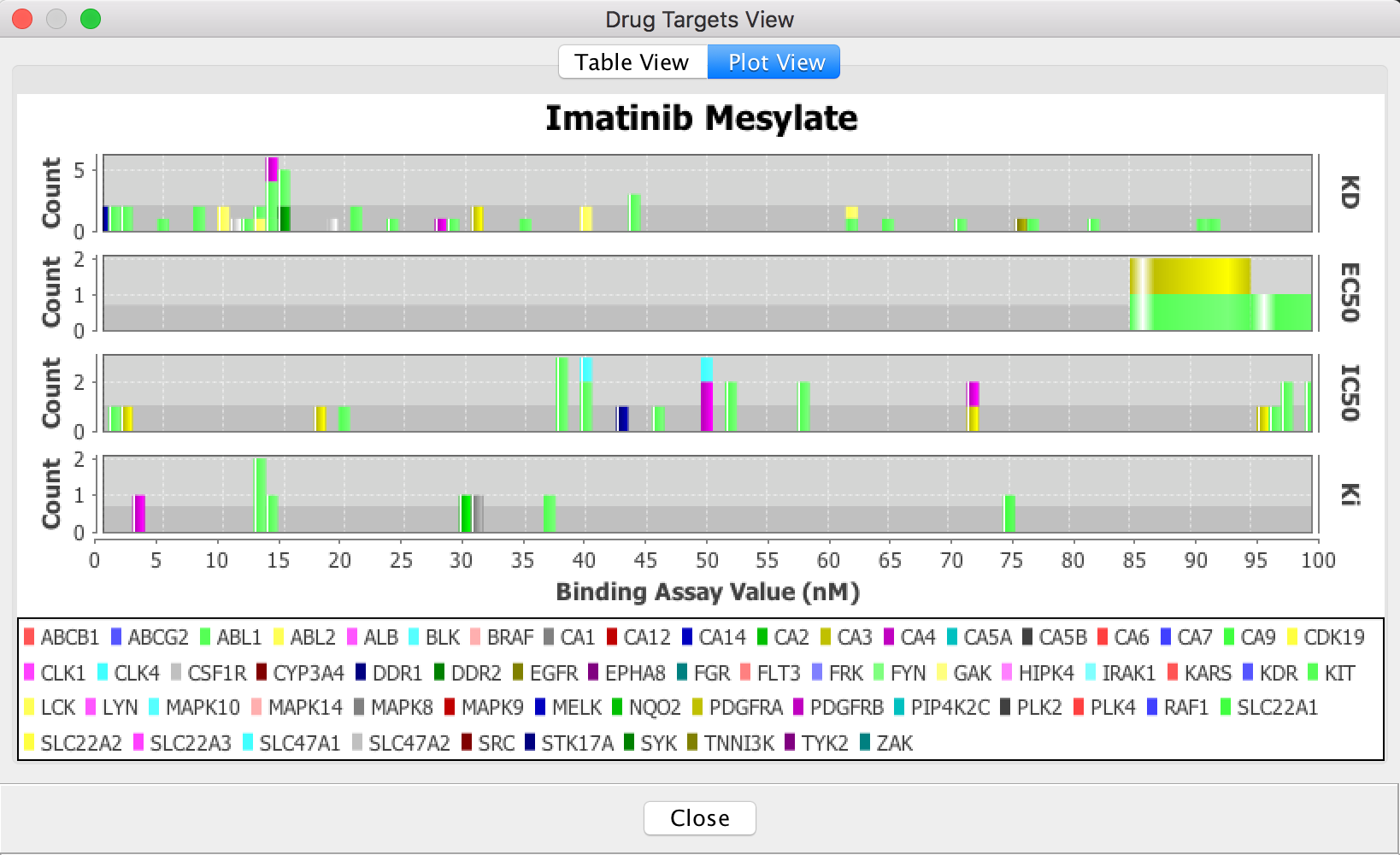 药物目标情节视图
药物目标情节视图 -
在路径图中查看癌症药物:使用中的弹出菜单“获取癌症药物”,可以将癌症药物直接覆盖到显示的路径图上通路关系图视图.获取的癌症药物显示在路径图中,与包含显示药物靶点的实体相链接。
 提取抗癌药物
提取抗癌药物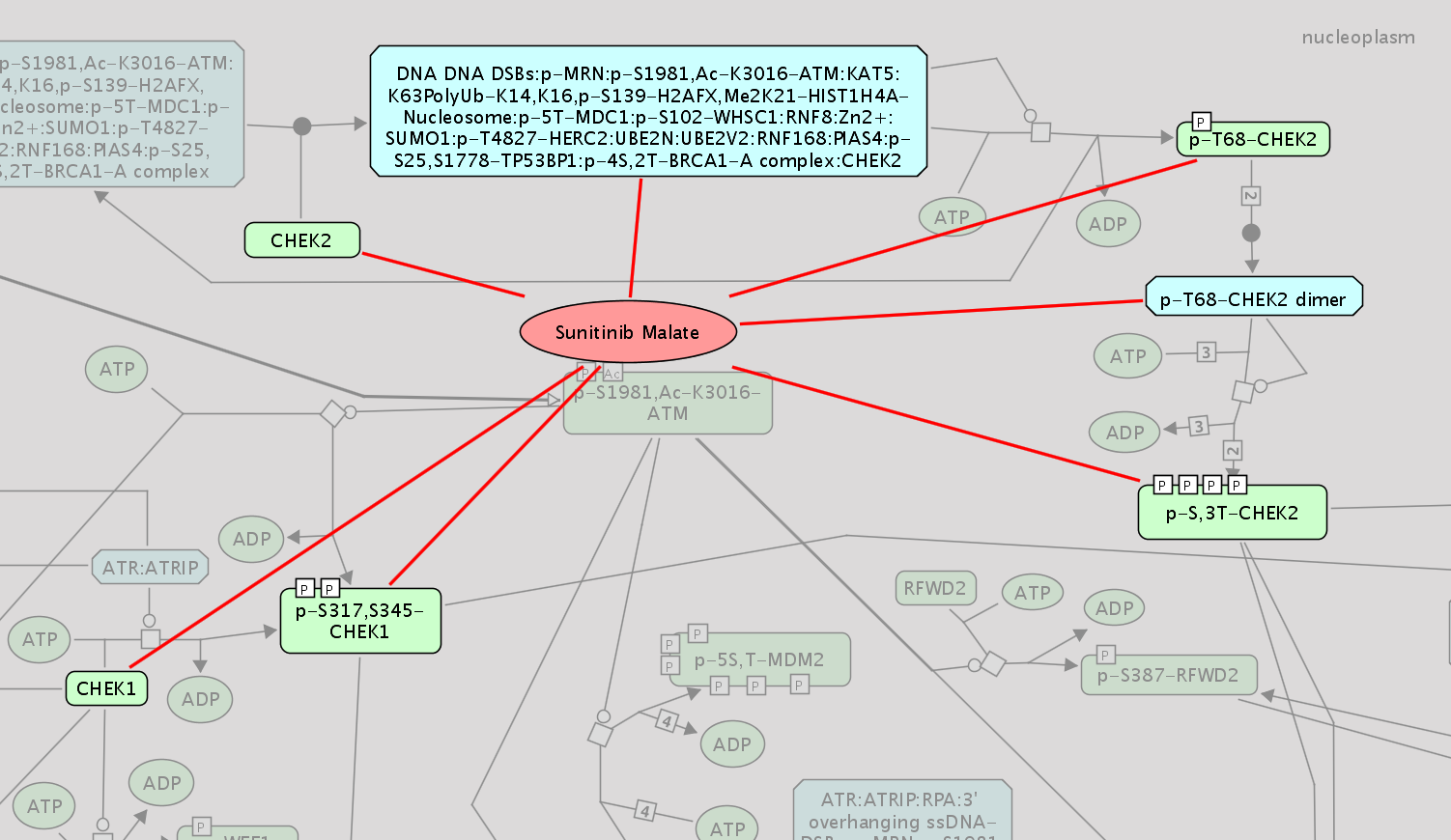 路径图中的药物注意:使用弹出菜单“Filter Drugs”调整过滤器,在路径图视图中选择药物和相互作用;使用“移除叠加的相互作用”来移除显示的药物;您可以在路径图中选择一个显示的实体,然后使用弹出菜单“获取癌症药物”只显示针对所选实体的药物。
路径图中的药物注意:使用弹出菜单“Filter Drugs”调整过滤器,在路径图视图中选择药物和相互作用;使用“移除叠加的相互作用”来移除显示的药物;您可以在路径图中选择一个显示的实体,然后使用弹出菜单“获取癌症药物”只显示针对所选实体的药物。
要查看显示的药物与其靶标实体之间相互作用的详细信息,请选择链接并使用pupup菜单“Show Details”打开药物靶标视图。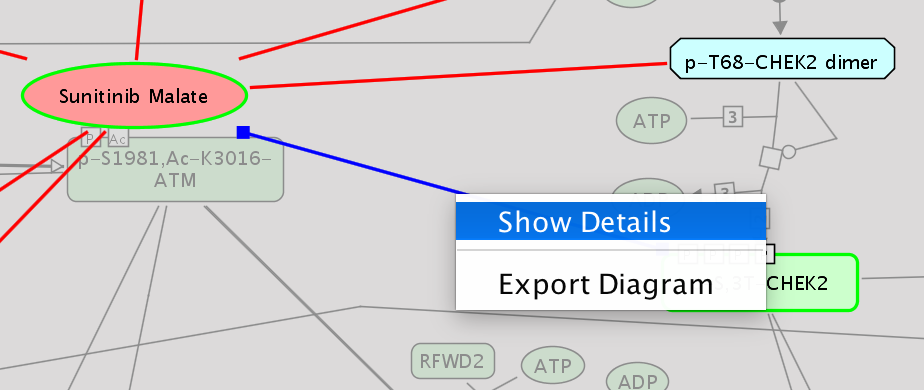 查看详细信息
查看详细信息 -
执行系统路径影响分析:在癌症药物列表表(见上面),您可以通过选择“运行路径影响分析”按钮,对所有具有实体级别视图的Reactome路径执行路径影响分析。分析完成后,影响结果显示在Cytoscape table Panel中所选药物的表格中。下面是伊马替尼的结果示例:
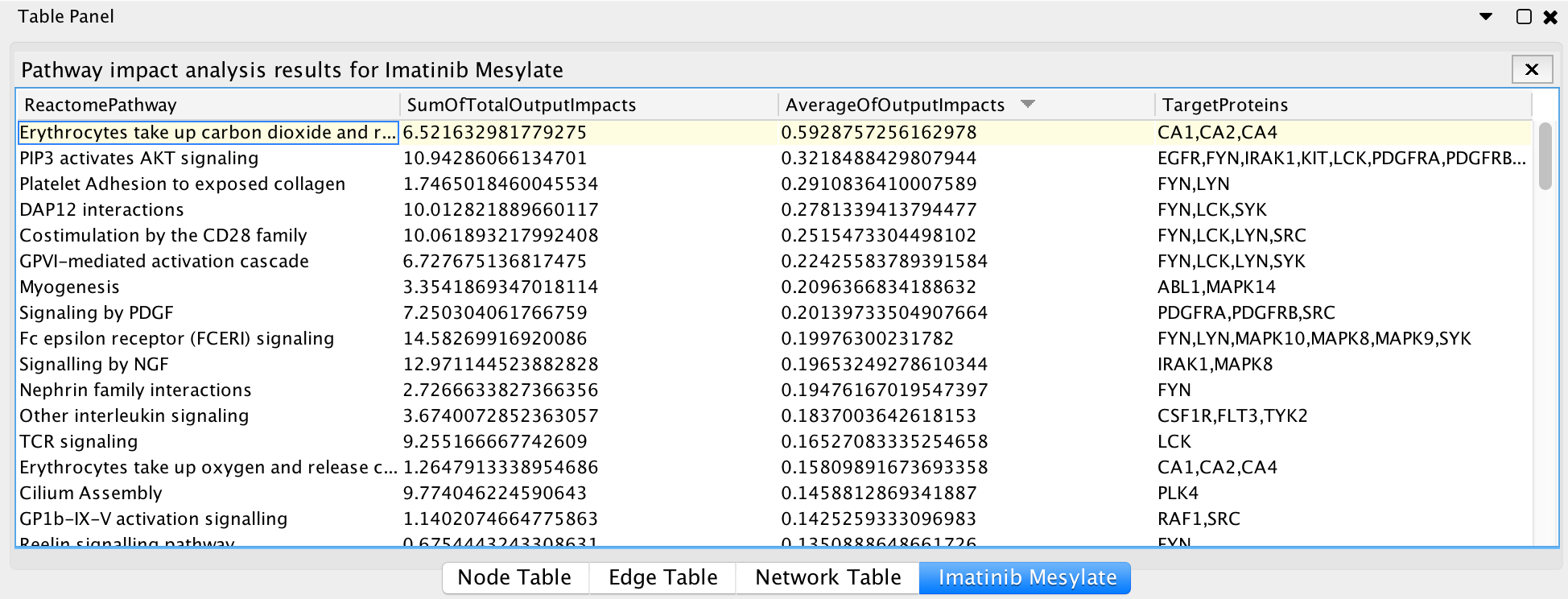 途径影响分析结果请注意:您可以使用POPUP菜单“在图中查看”打开路径图,并在结果表中使用菜单“导出表”将表导出到文本文件中。
途径影响分析结果请注意:您可以使用POPUP菜单“在图中查看”打开路径图,并在结果表中使用菜单“导出表”将表导出到文本文件中。










在FI网络中可视化癌症药物
Reactome FI网络提供了蛋白质/基因之间基于网络的视图,其中每个基因/蛋白质仅显示一次。在FI网络环境中可视化癌症药物显示了癌症药物与其靶点之间的简化关系。在网络视图,使用弹出菜单“Reactome FI/Overlay Cancer Drugs/Fetch Cancer Drugs”,加载FI网络中显示的蛋白质/基因的癌症药物。装载的药物以及药物与蛋白质/基因之间的相互作用分别呈现在绿色菱形和蓝色边缘。
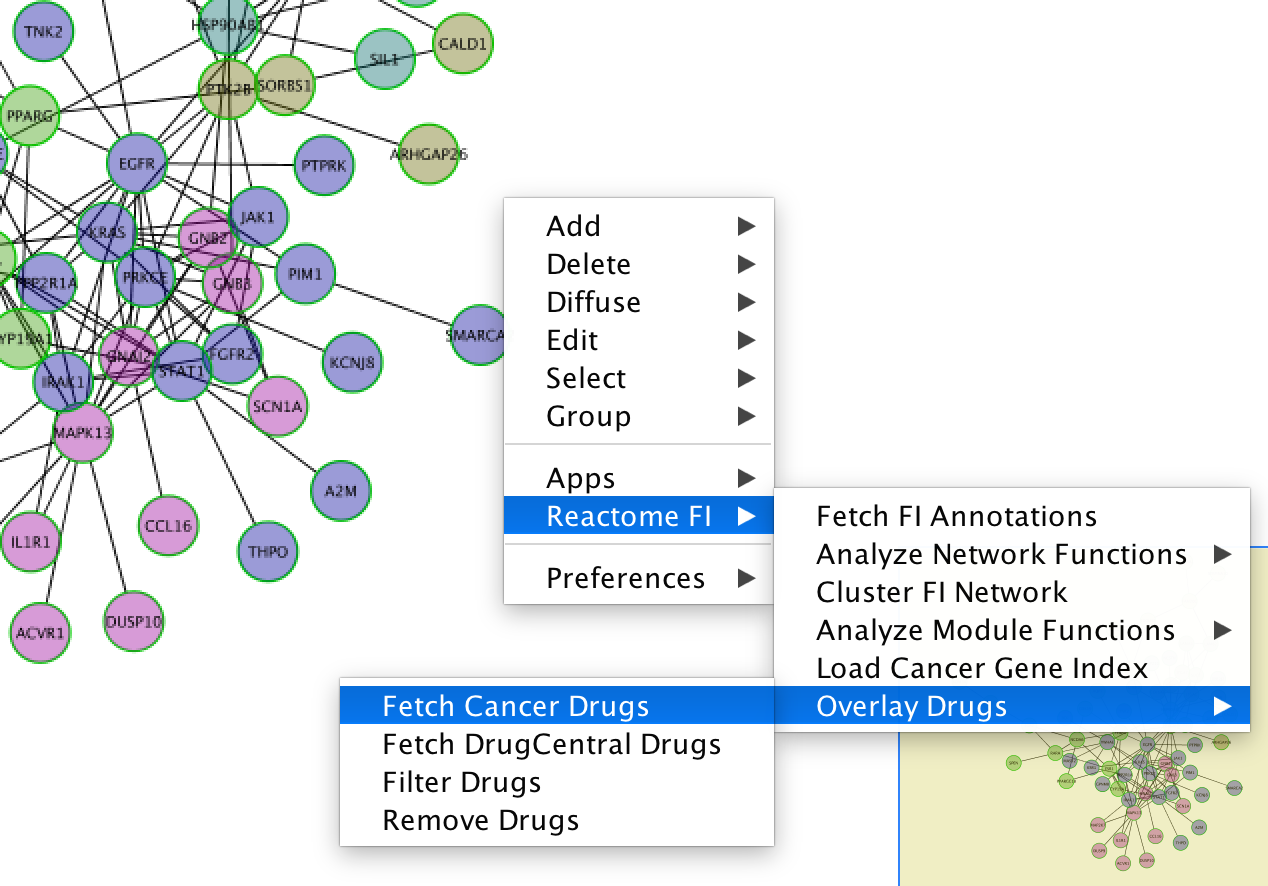
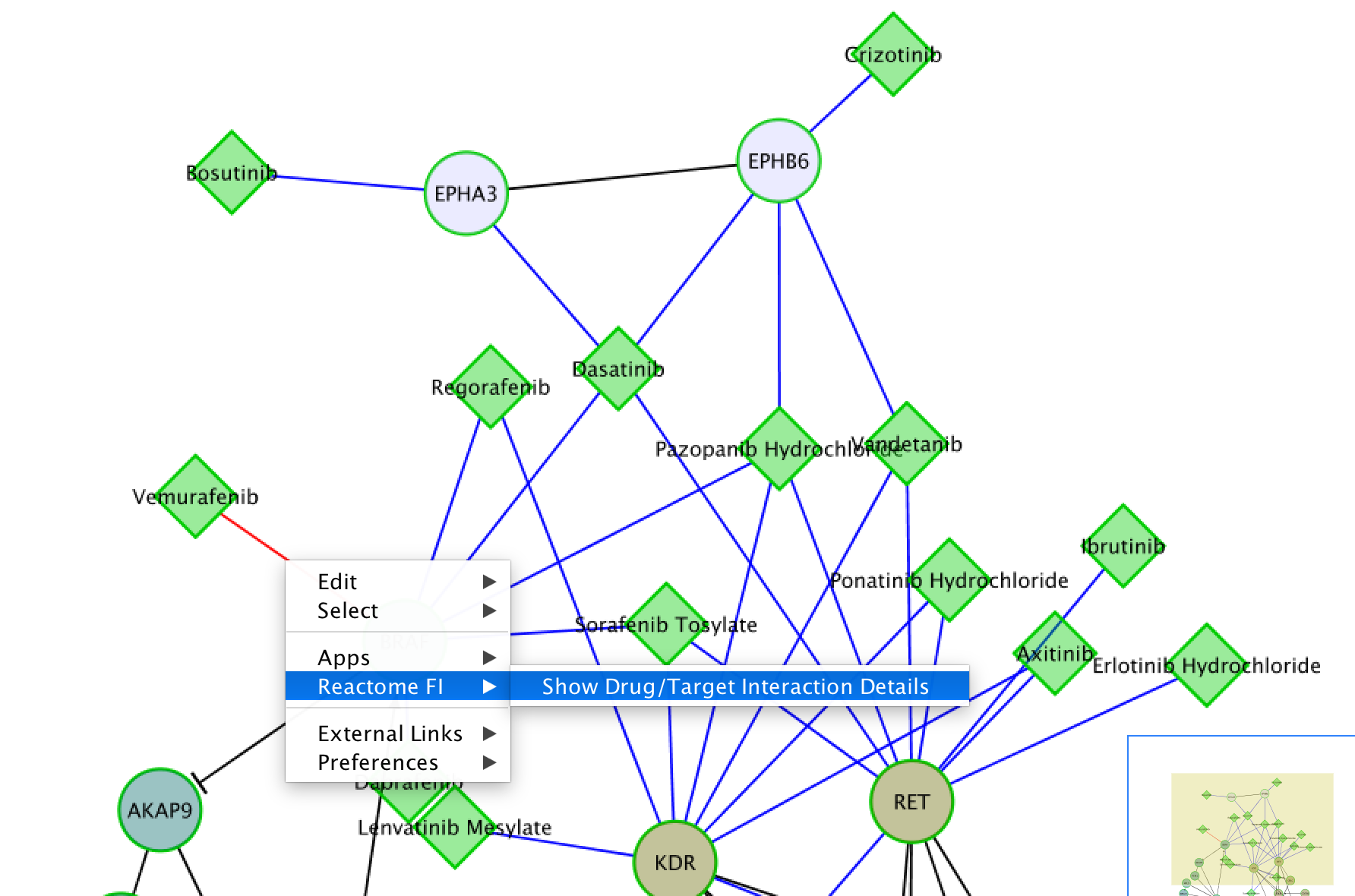
请注意:要移除FI网络视图中覆盖的药物,使用弹出菜单“Reactome FI/Overlay Cancer drugs / remove drugs”;就像在路径图视图中一样,你也可以通过使用“Filte Drugs”弹出菜单来应用过滤器;要查看药物和靶点相互作用的详细信息,请选择边缘,然后使用弹出菜单“Reactome FI/Show drug / target interaction details”。(您可能需要放大所选的边缘。)
想象药物中枢药物
在DrugCentral中收集的可视化药物与在cancer Targetome中收集的癌症药物类似。您可以使用弹出菜单“查看DrugCentral药物”路径树中列出所有药物DrugCentral数据库中收集,“获取DrugCentral药物”弹出菜单路径图视图中覆盖药物途径,或“ReactomeFI /覆盖药物/取回DrugCentral药物”覆盖药物到FI网络在网络视图。
模拟药物对通路活动的影响
将癌症药物覆盖到反应途径的背景下,其FI网络有助于用户了解应用药物对途径活动和网络行为的潜在影响。然而,途径上的药物的实际扰动可能更复杂。执行途径模拟可以帮助用户了解实际的影响。ReactomeFiviz实现功能以帮助用户执行基于布尔的网络的药物模拟。在做药物模拟之前,请阅读部分基于布尔网络的路径分析第一。在本节中,我们将使用路径HDR通过同源重组(HR)或单链退火(SSA)以药物伊马替尼为例(注:要获得以下截图,请打开此通路的图表,然后使用名称过滤器获取癌症药物并将药物过滤到伊马替尼)。
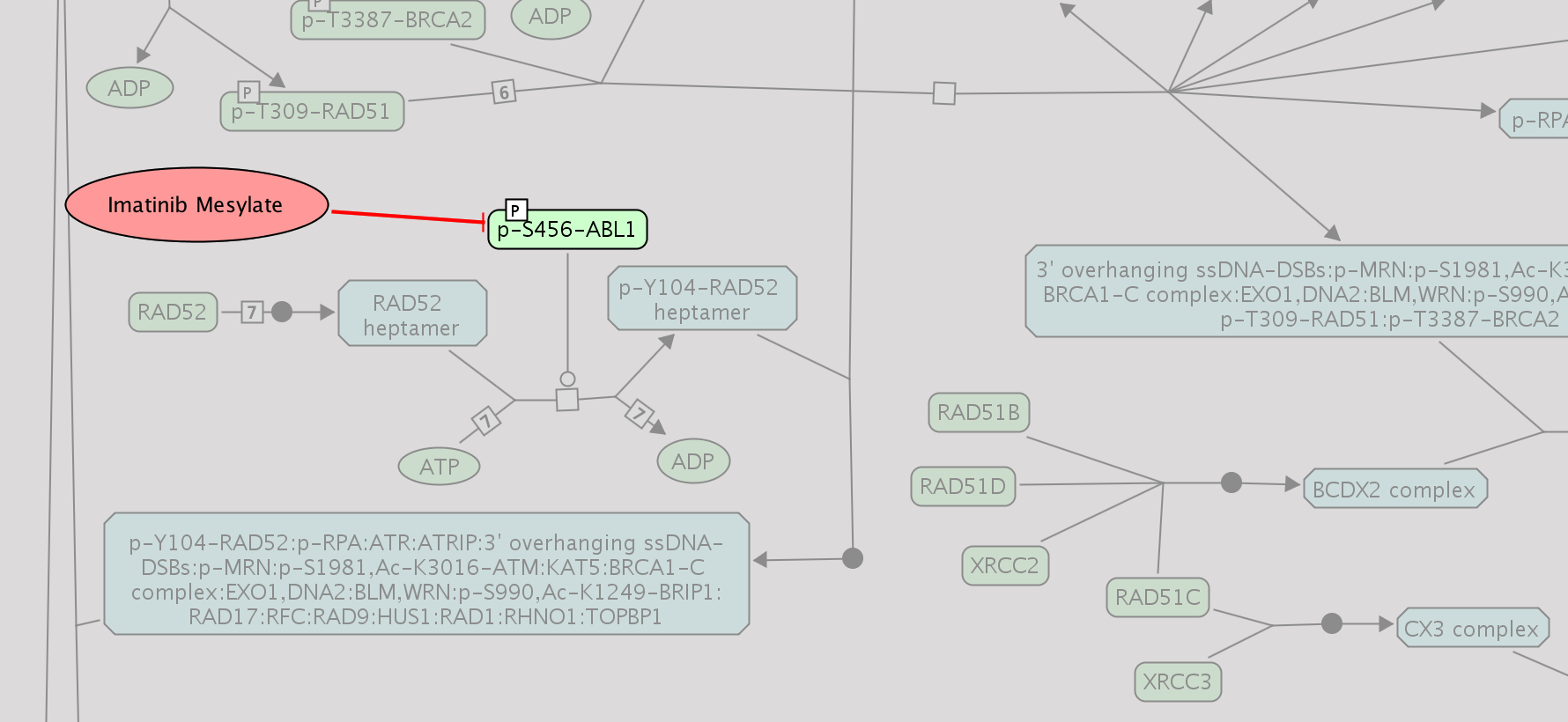
- 建立新的药物模拟系统:在路径图视图中,使用弹出菜单“运行逻辑模型分析”。在新建仿真对话框中,输入仿真的名称(例如Imtatinib)和默认值,然后选择与门的模式,就像在常规逻辑模型仿真中那样。要执行药物模拟,选择药物应用选项卡和药物数据源,然后单击“…”按钮,弹出药物选择对话框。
请注意:您需要调整药物过滤器,以显示伊马替尼的所有靶标,如下面的截图所示。根据收集到的抗癌药物与其靶标之间相互作用的注释,将选择默认的修饰类型(正如预期的那样,大多数是抑制)。根据我们聚合的药物/目标数据库中收集的亲和性,预配置修饰强度。
 伊马替尼BN模拟
伊马替尼BN模拟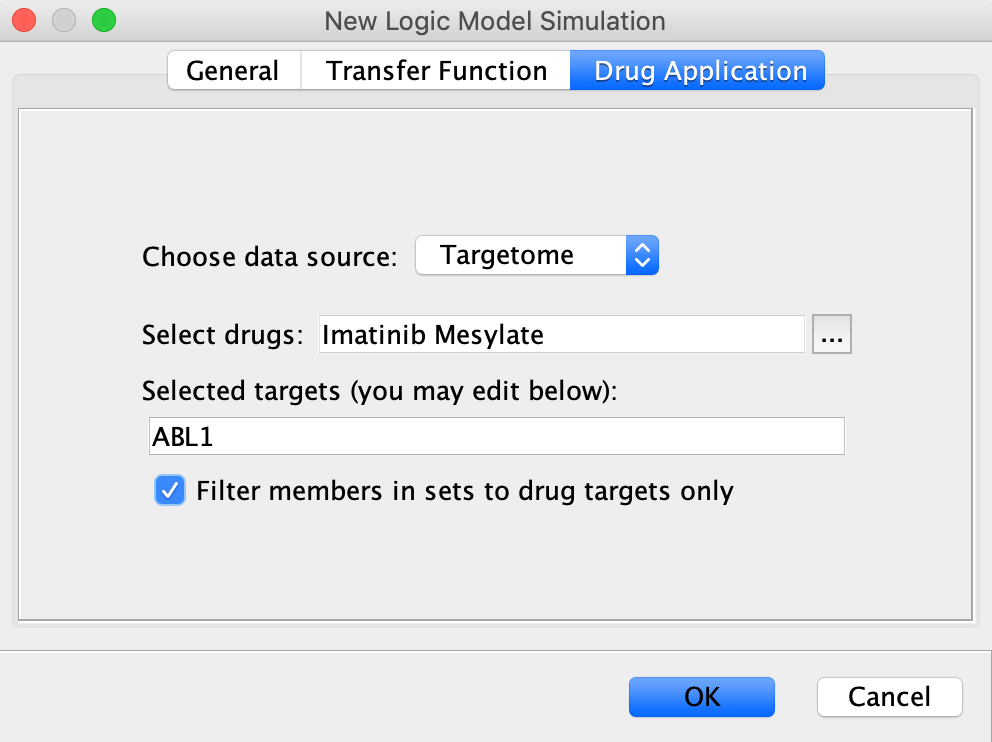 伊马替尼BN模拟2
伊马替尼BN模拟2
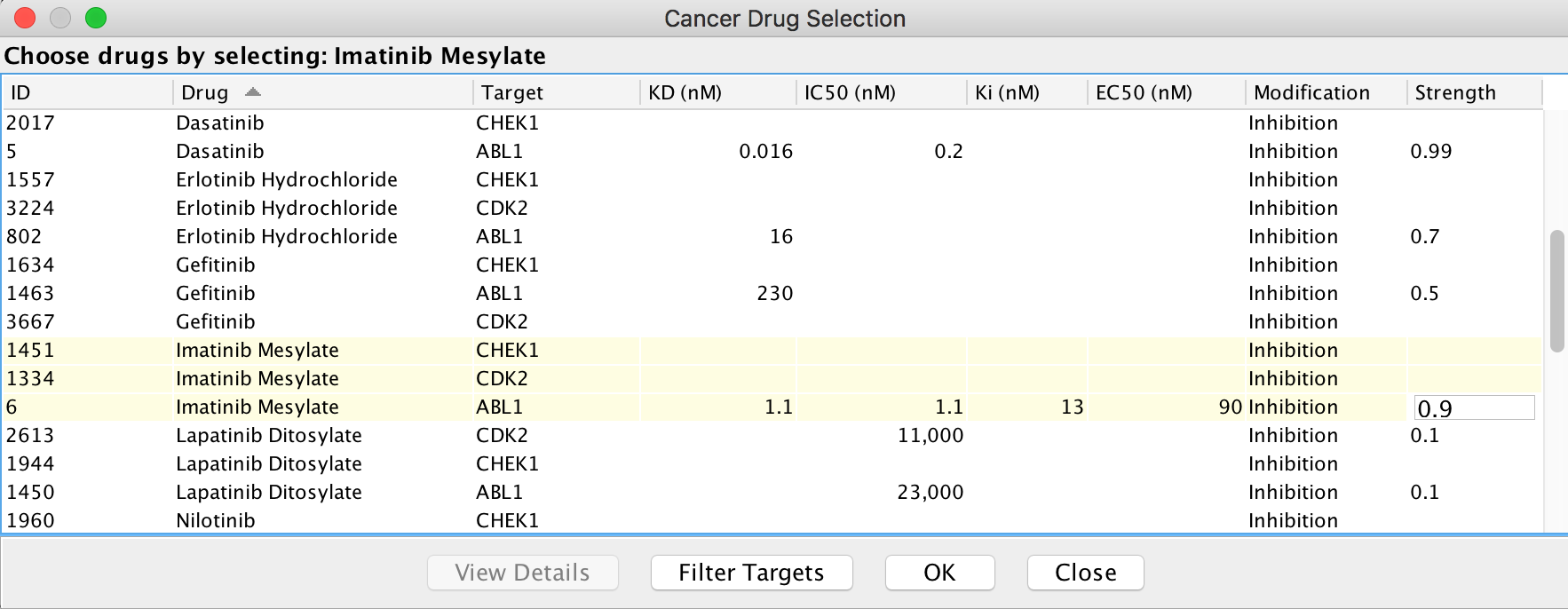 选择BN仿真的药物
选择BN仿真的药物
注意:由于伊马替尼与CHEK1或CDK2之间的相互作用没有亲和性,CHEK1和CDK2在新模拟对话框中没有列出。勾选“Filter members in sets to drug targets”将选择EntitySet实例中被drug靶向的成员,强制显示药物引起的潜在路径影响。 - 用药物进行模拟:药物选择对话框中的药物配置将复制到结果面板中显示的布尔网络配置表中。单击“模拟”按钮执行模拟。模拟完成后,路径图中的实体将根据吸引子中的值以不同颜色高亮显示,详细的时间值显示在底部表格面板的表格中。
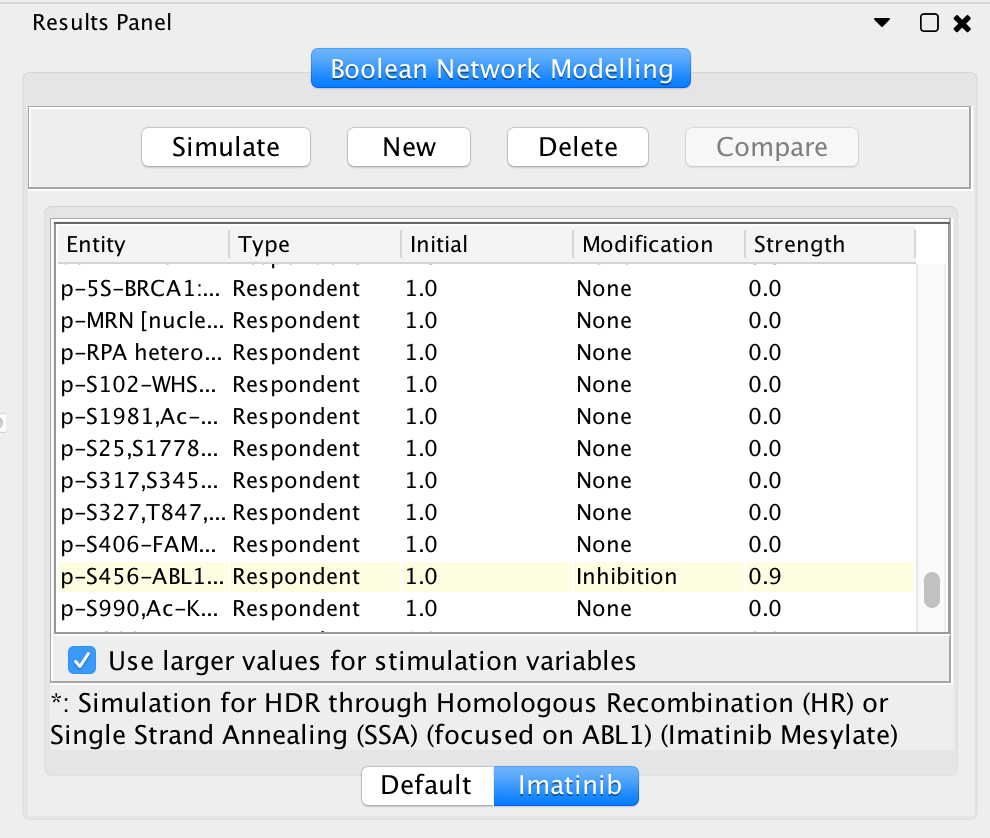 伊马替尼BN设置
伊马替尼BN设置
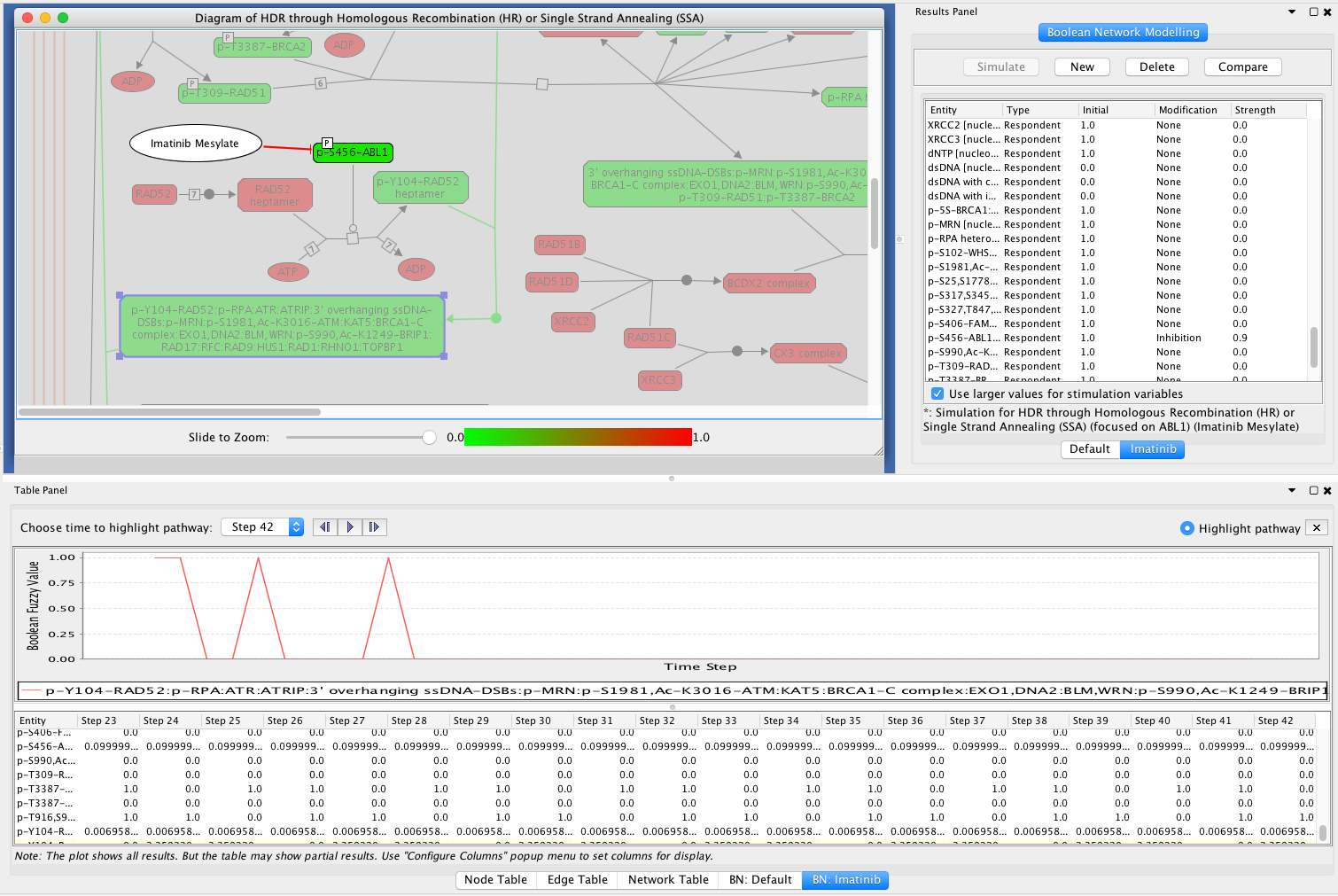 imatinib bn结果
imatinib bn结果 - 研究药物对通路活性的影响:要查看药物对通路活动的影响,在不应用癌症药物的情况下执行另一个布尔网络模拟(此处为默认值)(详情请参见基于布尔网络的路径分析).“HDR through homoologous Recombination (HR) or Single Strand退火(SSA)”路径默认初始配置的逻辑模型仿真结果截图如下:
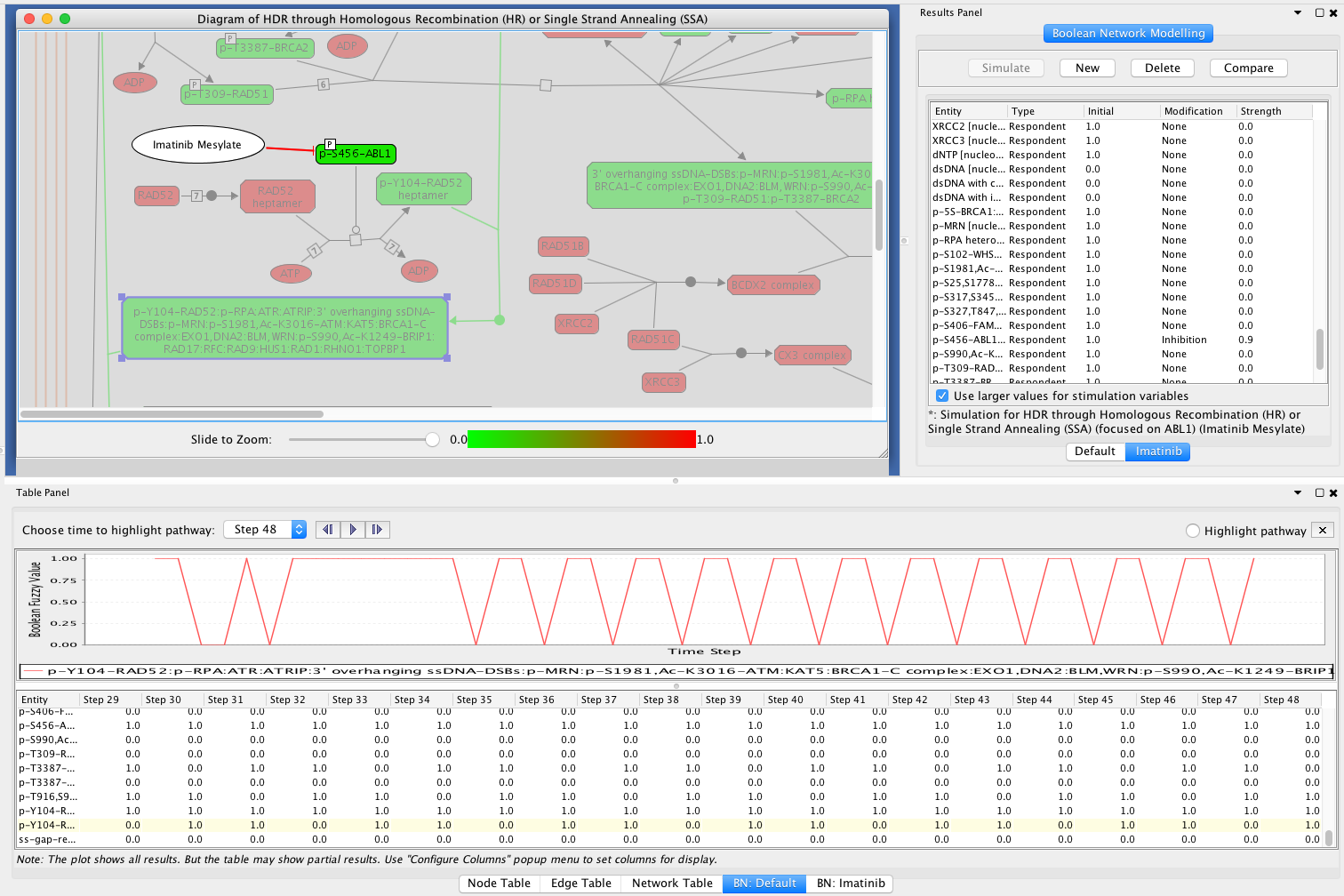 HDR通过HR或SSA默认结果
HDR通过HR或SSA默认结果
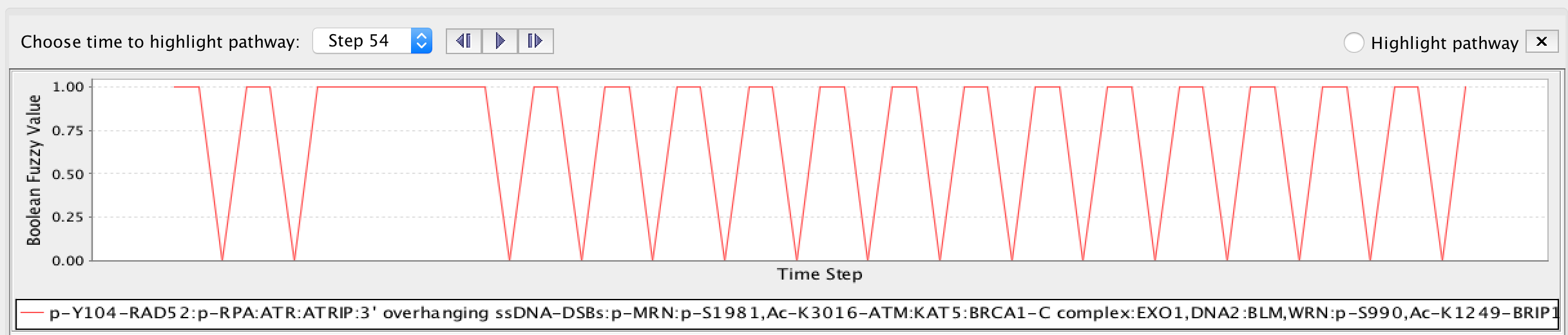
要查看药物对路径中显示的实体活动的影响,请选择该实体并在BN:Default和BN:Imatinib表中检查其时间行为。例如,下面选择了与ABL1相关的复合体(见上面的屏幕截图)(应用伊马替尼时向上,默认无药物时向下):
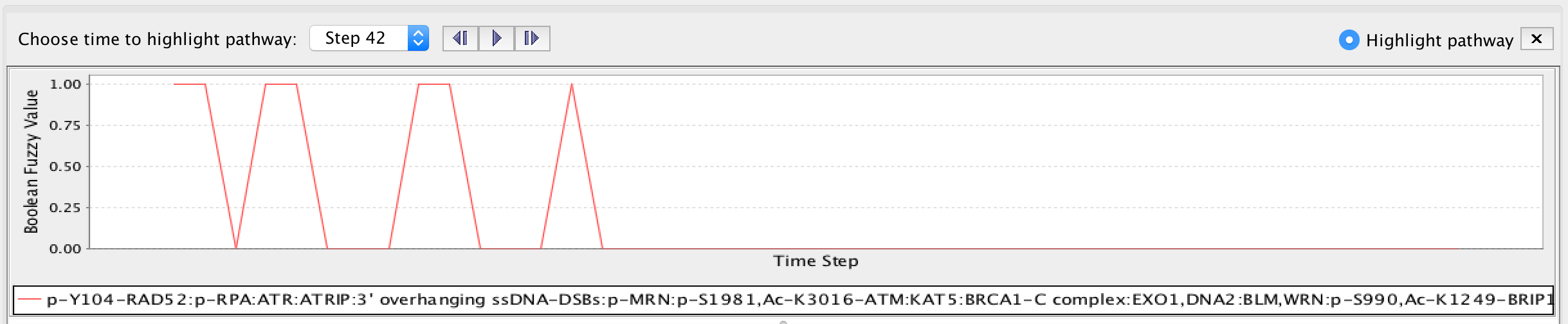 伊马替尼一个变量
伊马替尼一个变量
 默认的一个变量
默认的一个变量
你也可以使用“比较”按钮来检查从两个模拟中计算出的吸引子的详细差异。
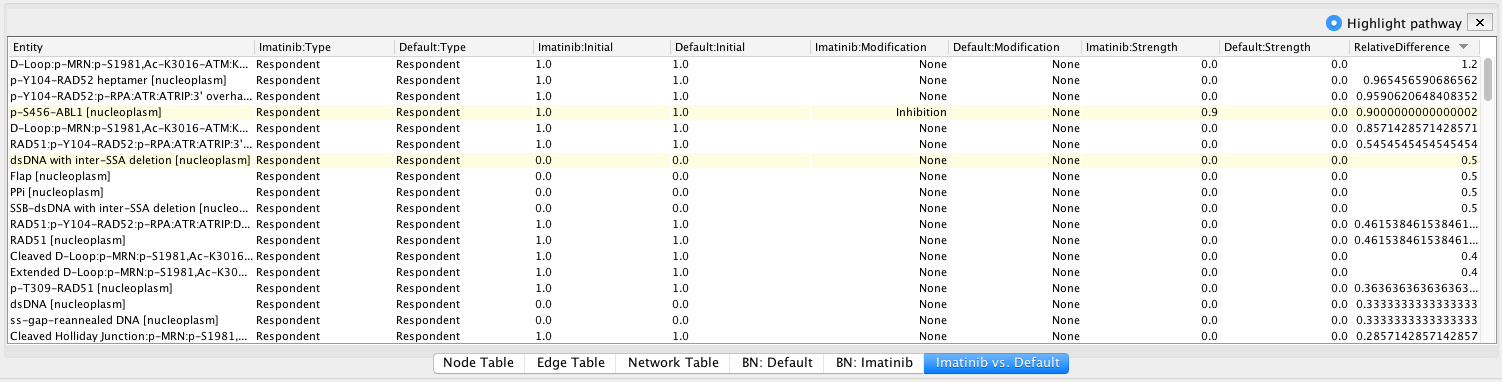 p-S456-ABL1值
p-S456-ABL1值
请注意:通过以上对比,我们可以看出,伊马替尼的应用将显著影响HDR复合物的形成,伊马替尼对DNA修复通路的影响已经有报道(如:伊马替尼(STI571)在表达BCR/ abl的白血病细胞中诱导DNA损伤,但在正常淋巴细胞中不诱导DNA损伤).由于新版本的ReactomeFiviz中的路径注释更新,您可能会看到一些不同的模拟结果。









执行scRNA序列数据分析和可视化
ReactomeFIViz为用户实现了一套功能,用于进行scRNA序列数据分析和可视化。为此,我们将几个为scRNA seq数据分析和可视化开发的Popullar Python包打包到一个Python独立应用程序中。这些方案包括稀疏的用于常规SCRNA-SEQ数据分析和可视化和scVelo用于基于RNA速度的数据分析和可视化。
请注意:对于scRNA seq数据分析和可视化,您需要在计算机上安装Python 3.7。如果尚未在计算机上安装Python,可以通过从下载安装程序来完成https://www.python.org/downloads适合您的电脑。我们仅测试了Python 3.7,因此建议您使用3.7来进行这些功能。但是,您无需从ReactomeFiviz轻松地安装我们的独立Python应用程序。当需要时,只要您指向正确的Python应用程序路径(即目录和应用程序文件),ReactomeFiviz将自动下载并更新您的应用程序。
标准分析通过Scanpy
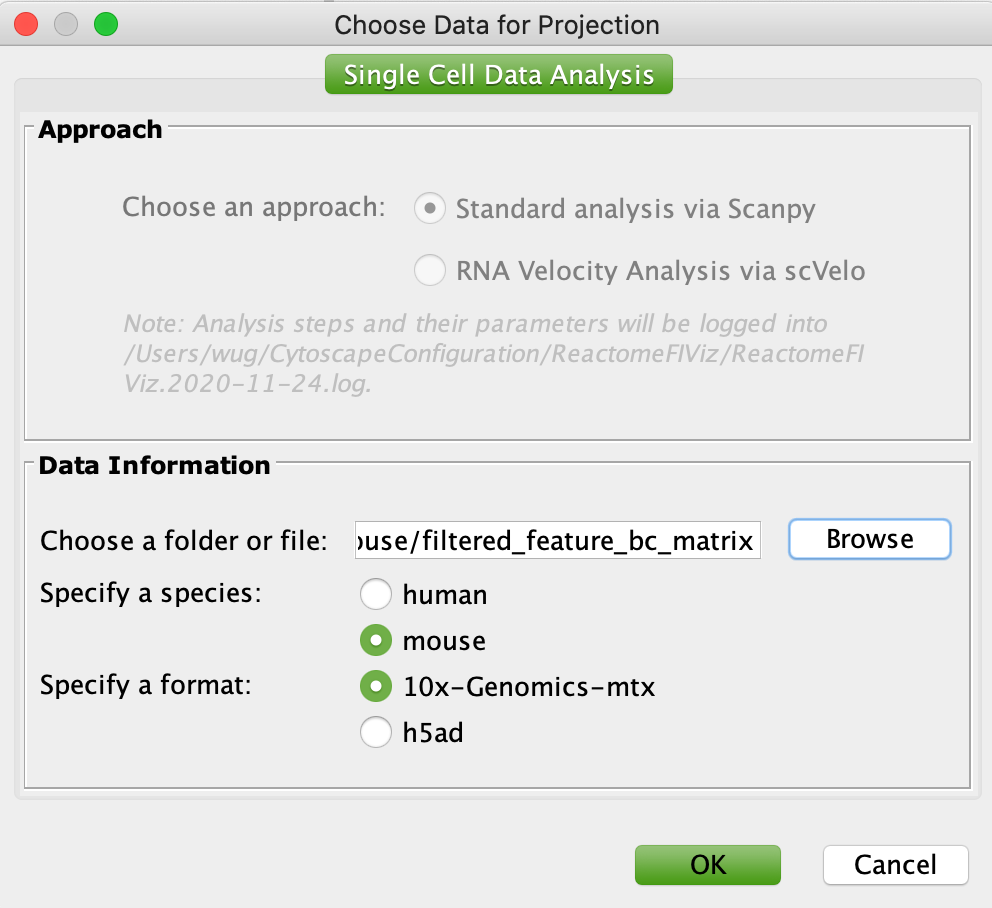
- 建立分析:Python包,稀疏的,为scRNA-seq数据提供了一组强大的分析和可视化功能。ReactomeFIViz包装了这些功能,用户可以利用Cytoscape和ReactomeFIViz提供的途径和网络分析和可视化设施。要使用scanpy进行scRNA-seq分析,选择菜单Apps/Reactome FI/Single Cell analysis /Analyze得到如下所示的配置窗口:ReactomeFiviz支持从鼠标和人类生成的SCRNA-SEQ数据。您应该为您的数据和格式选择物种。如果您的数据处于10x-genomics-mtx格式,则应选择包含该格式的文件的目录。您可以检查一个估算方法。目前,ReactomeFiviz支持魔术只进近。您还可以检查总计数和/或pct计数以确定回归了控制不需要的变化。
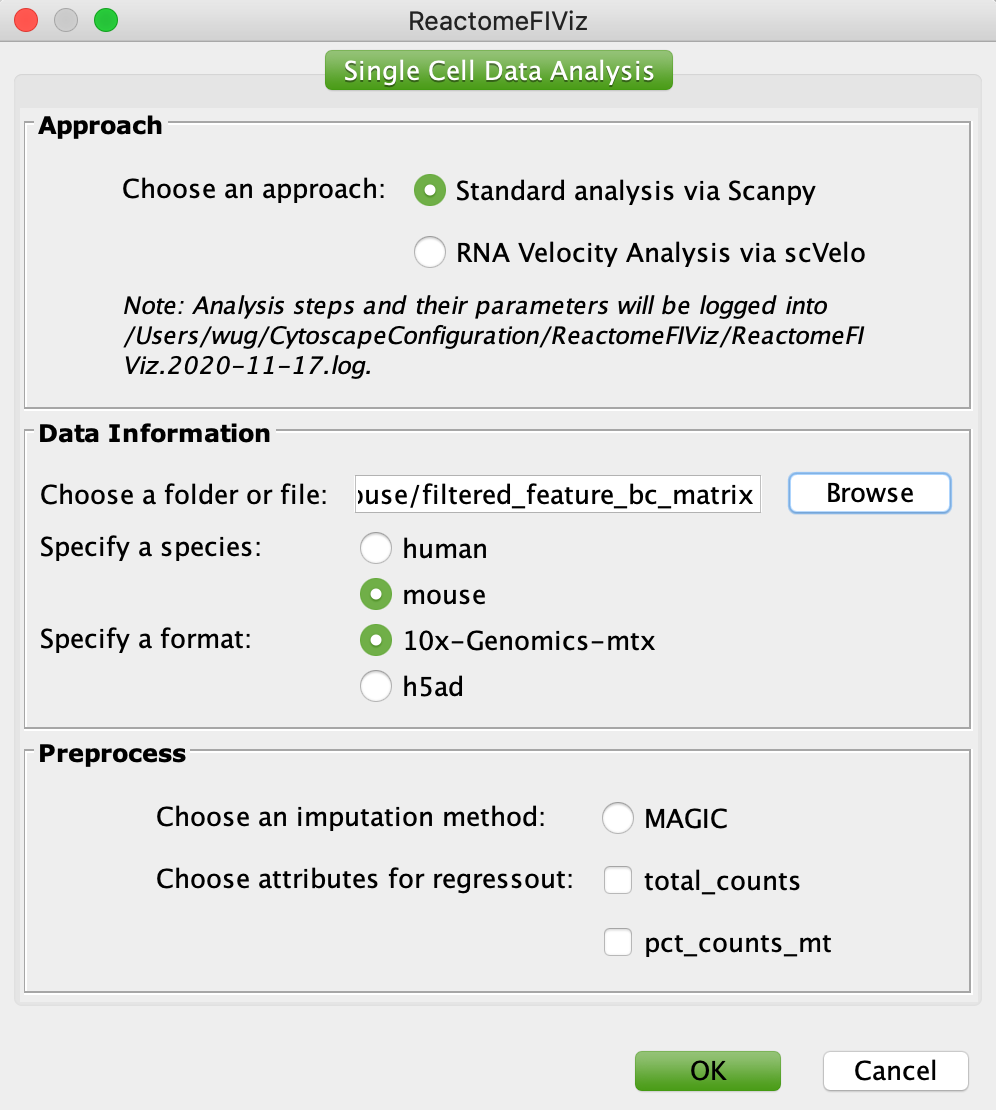 scRNA序列分析配置
scRNA序列分析配置
请注意:所有的分析步骤及其参数都被记录到您的用户文件夹中的CytoscapeConfiguration/ReactomeFIViz/ReactomeFIViz.{date}.log中以供查看。 - 为ReactomeFIViz配置Python:如果你还没有这样做,当ReactomeFIViz下载用于scRNA-seq数据分析和可视化的ReactomeFIViz Python应用程序时,会要求你使用下面的配置对话框为ReactomeFIViz设置Python。请注意当前只支持Python 3.7。
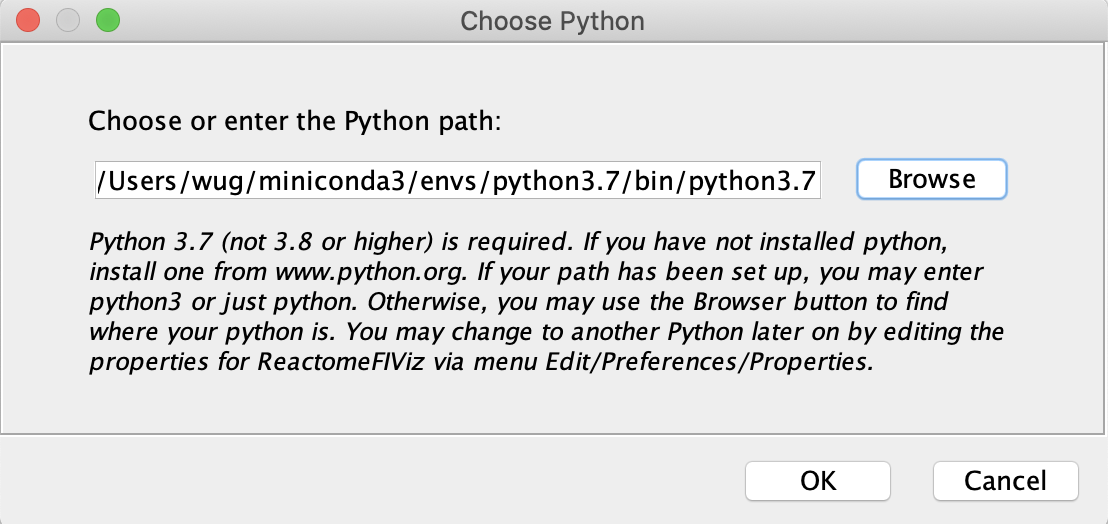 设置Python
设置Python
ReactomeFIViz使用scanpy提供的功能进行预处理、归一化、UMAP分析、细胞聚类和所有其他scRNA-seq分析,但归一化除外魔术如果勾选此项。详细信息见scanpy文档:https://scanpy.readthedocs.io/en/stable/api/index.html.对于用于这些函数的参数,打开ReactOmeFiviz。{data} .log文件(见上文)。 - Visuzlize手机网络:根据样本大小和计算能力,完成分析可能需要几分钟。然后,两个网络(一个用于细胞簇,另一个用于单个细胞)将显示在Cytoscape中,并分别列在左侧网络选项卡的“SingleCellClusterNetwork”和“SingleCellNetwork”下。请注意您可以使用内置的Cytoscape风格特性和其他配置属性来调整这两个网络的渲染。通过单击菜单“帮助/用户手册”,查看Cytoscape的用户手册。要在网络中显示或隐藏边缘,使用弹出菜单Reactome FI/ show edges(见下面的截图)。集群网络中的集群名称是根据集群中细胞数量排序的集群的秩来命名的。例如,cluster0具有最大的单元数。
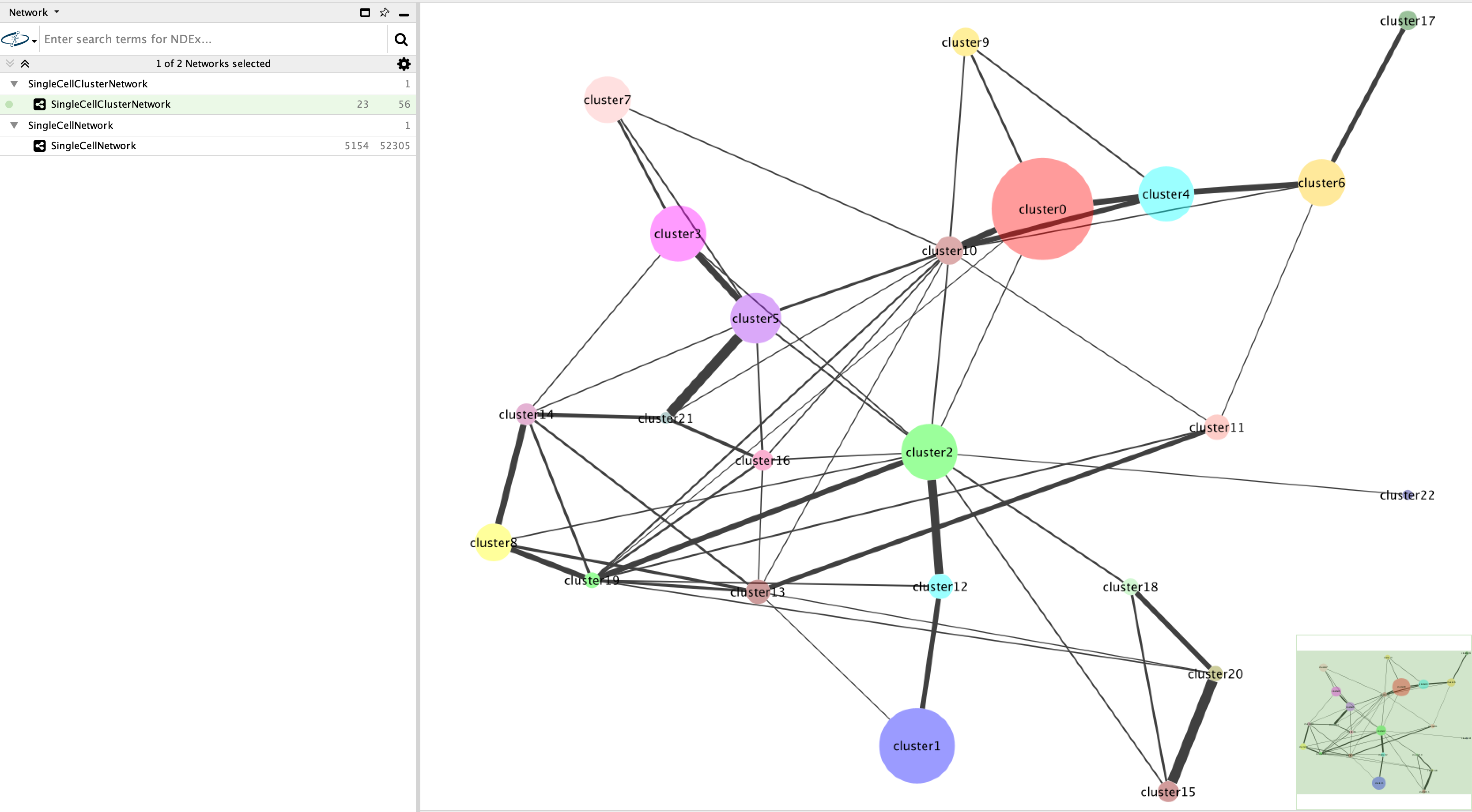 ScRNA-seq集群网络
ScRNA-seq集群网络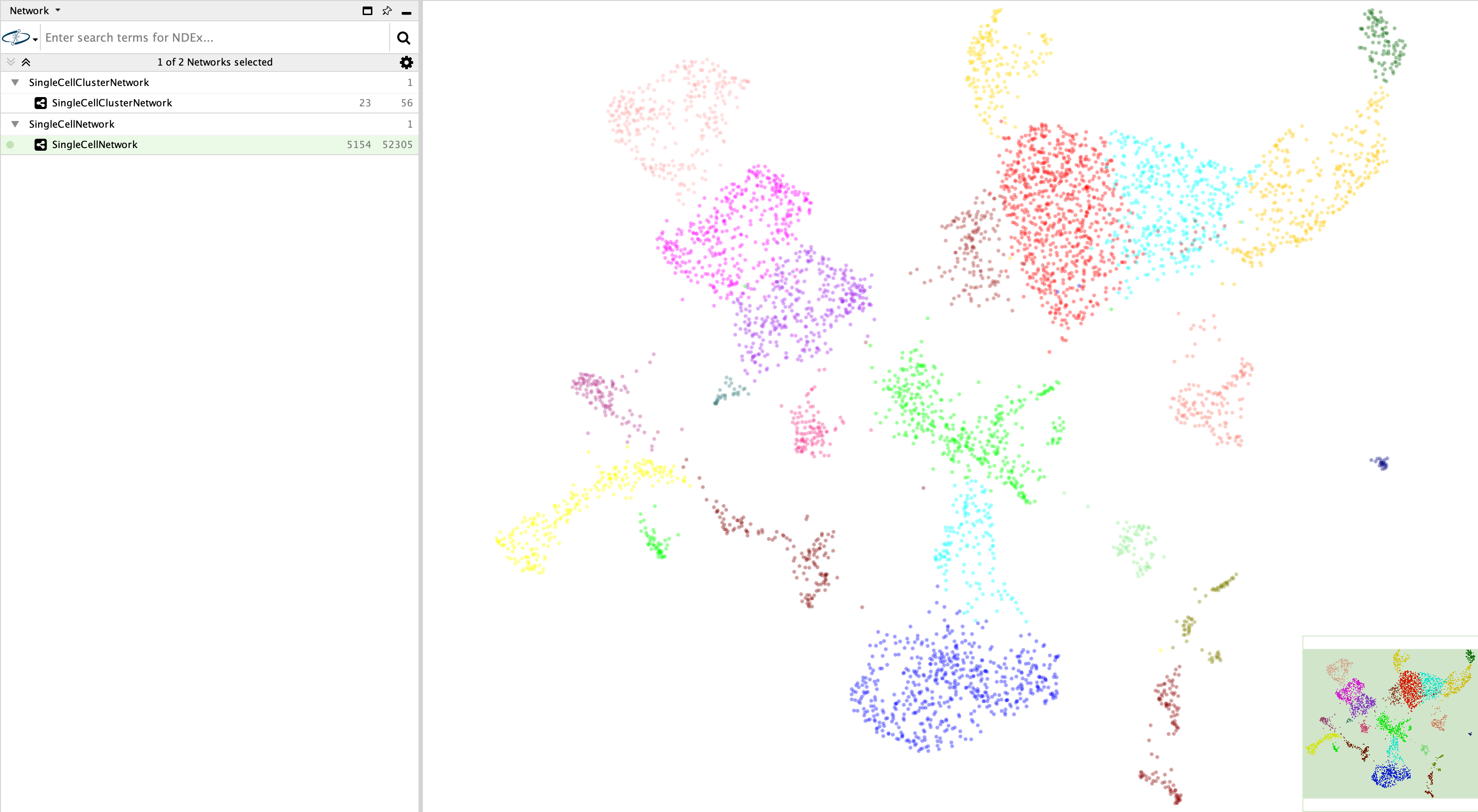 ScRNA-seq蜂窝网络
ScRNA-seq蜂窝网络 - 分析scRNA-seq数据要探索加载的scRNA-seq数据并进行进一步的分析,您可以使用cell cluster或single cell network视图中提供的弹出菜单,如下所示:
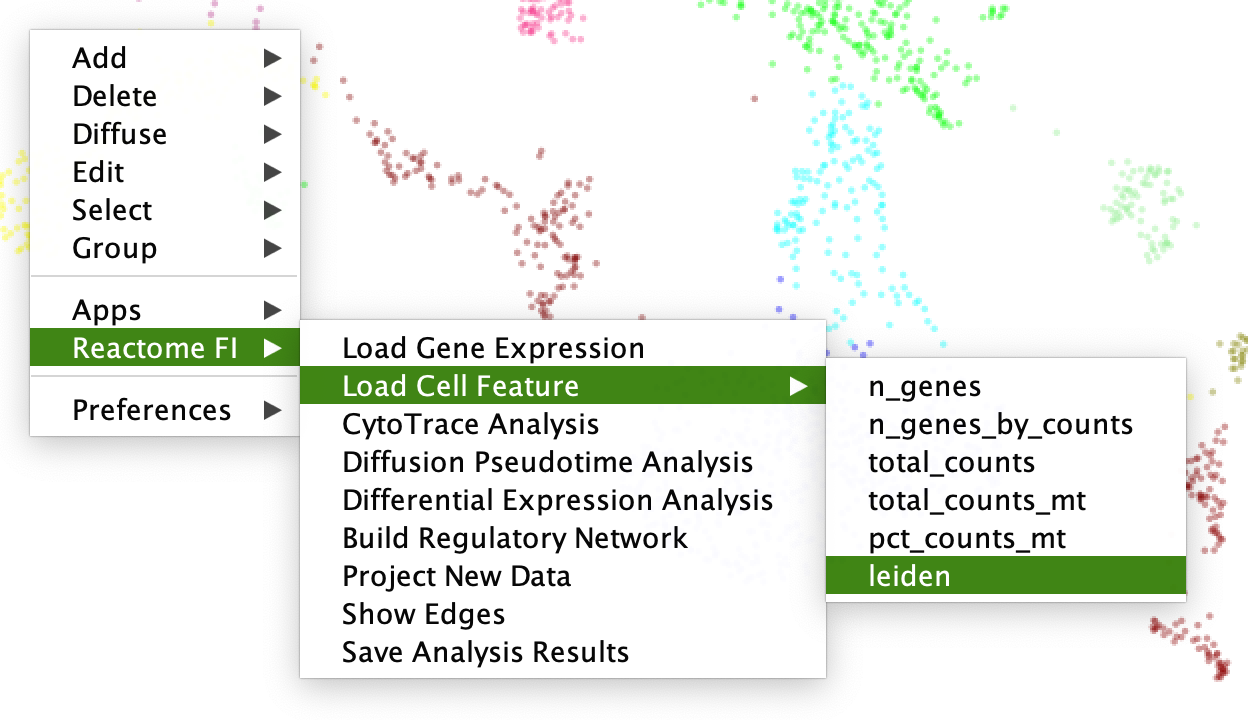 SCRNA-SEQ标准分析弹出菜单
SCRNA-SEQ标准分析弹出菜单- 负载基因表达:将基因的表达值叠加到网络上。点击此菜单后,您可以在输入对话框中输入基因名称。
请注意:细胞集群使用集群中细胞的中位数为基因表达和细胞特征着色(下图)。 - 负载细胞特性:通过选择子菜单覆盖细胞特征,例如,n_genes(总基因),n_genes_by_counts(总基因有计数),total_counts, total_count_mt(线粒体基因),pct_counts_mt(线粒体基因百分比),和leiden(基于莱顿算法)。
请注意:细胞簇网络和单细胞网络在分析后渲染时,都是根据莱顿聚类结果着色的。要回到原来的颜色,请选择负载单元功能/莱顿。 - 细胞追踪分析:根据每个细胞检测到的表达基因数量,进行细胞trace分析,预测细胞的差异状态。ReactomeFIViz提供了基于发布于https://cytotrace.stanford.edu.关于CytoTrace的详细信息,见论文原文:单细胞转录多样性是发育潜力的标志.你可以在CytoTrace的网站上找到更多信息:https://cytotrace.stanford.edu.
请注意:分析可能需要几分钟。分析后,将根据预测的差异状态值对单元格进行着色,最大差异状态值(黄色)为0到1:0,最小差异状态值(蓝色)为1。分析结果缓存在ReactomeFIViz中,并在名为“cytotrace”的新列中列出在底部的节点表中。当您在分析后再次选择此菜单时,结果将被覆盖,而不执行其他分析。名为“cytotrace”的新菜单项也将添加到“称重传感器功能”弹出菜单中,以便您可以直接加载这些结果。
请注意:在分析之后,您可能不会看到这个菜单项。尝试切换到另一个网络视图,然后返回来刷新菜单项。 - 扩散伪时间分析(DPT):基于网络扩散进行单元轨迹推断。有关该算法的详细信息,请参阅scanpy.tl.dpt及原始论文:PAGA:图抽象通过单个单元的拓扑保持图来协调聚类和轨迹推断.为了进行这种分析,需要一个细胞的id,它应该被视为轨迹的根。如果你知道这个单元格是什么,你可以直接在下面的对话框中输入它。如果您不知道根是什么,但知道根可能驻留在哪个集群或集群中,则可以在第二个文本字段中输入集群。ReactomeFIViz将尝试根据您指定的集群为您推断一个可能的单元根PageRank.关于单元根推理算法的详细信息,请参见infer_cell_root.请注意:如果你不知道细胞根是什么,你可以尝试几种方法。如果您已经进行了CytoTrace分析,您可以选择具有最大的CytoTrace值的单元格作为单元格根。你也可以尝试有最大数量的检测基因作为根进行探索数据分析。要选择用于推断细胞根的细胞簇,应该选择具有最大的CytoTrace值或基因数的细胞簇。如果您在上面对话框中的两个文本字段中都输入了值,那么第一个文本字段中的值将被用作单元格的根。
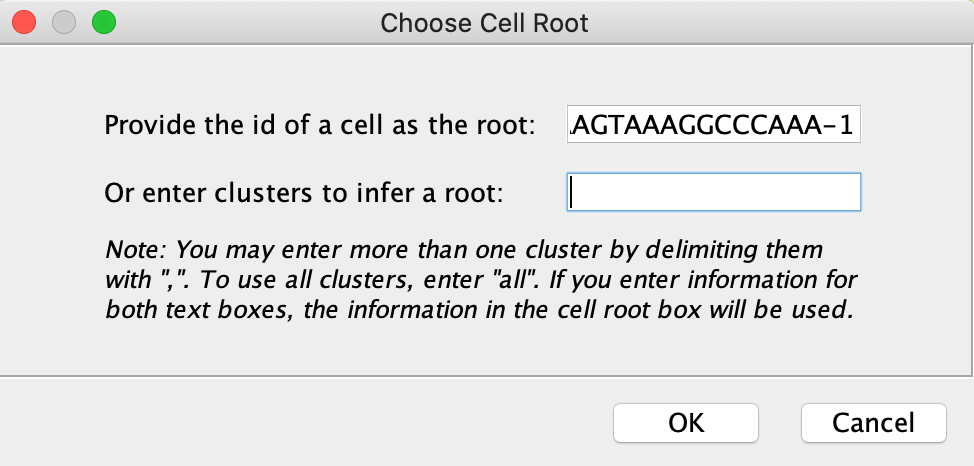 配置DPT的Cell Root
配置DPT的Cell Root
DPT分析完成后,将根据DPT值(0 ~ 1)对单元格进行着色,其中0为轨迹中最早的单元格(黄色),1为最近的单元格(蓝色)。一个名为“dpt_pseudotime”的新列将被添加到底部的节点表中,而“dpt_pseudotime”将被注册为“Load Cell Feature”弹出菜单下的新项,以便加载,而无需重复分析。
请注意:在分析之后,您可能不会看到这个菜单项。尝试切换到另一个网络视图,然后返回来刷新菜单项。 - 差异表达分析:使用细胞簇(组)和另一个细胞簇或所有其他细胞集群之间的差异基因表达分析t-test_overestim_var..要进行此分析,首先需要选择两组单元:基于聚类结果进行分析的一组单元和作为参考的另一组单元。您可以选择其他单元集群或所有其他单元作为参考。差分表达式分析结果如下表所示。你可以通过点击“添加”按钮选择一个或多个筛选器来筛选表中显示的基因。点击“Build FI network”按钮,为表格中显示的筛选基因创建一个FI网络。要进行途径富集分析,可以选择Binomial_test或GSEA.Binomial_test将使用筛选后的表中显示的基因,而GSEA分析将使用按分数排序的基因,包括表中没有显示的基因。
 选择用于不同的表达分析的单元组请注意:由所选基因构建的FI网络中的基因根据基因评分进行着色。有关如何使用FI网络的特性的更多信息,请参见基因设置/突变分析.当显示网络视图时,您可以打开路径图。要回到以前的网络视图,请关闭所有显示的路径图,然后在左侧控制面板的“网络”选项卡中选择网络。根据黑豹的同源映射文件,从人类路径预测小鼠路径。更多细节,请参阅:在Reactome中的推断事件.利用所提供的映射文件,从人体功能交互网络中预测出鼠标人体功能交互网络MGI,使用此链接下载:http://www.informatics.jax.org/downloads/reports/HOM_MouseHumanSequence.rpt.一个人类基因可以映射到多个老鼠基因上。因此,小鼠FI网络可能显示了一个带有多个小鼠基因注释的节点。例如,请参见下面的内容:原始的人类KLK3被映射到Klk1b9、Klk1b21和其他许多节点,它们都列在底部的节点表中,并在网络视图中作为该节点的标签。目前连接到FI网络中的这些节点指向人类基因(如GeneCard)。
选择用于不同的表达分析的单元组请注意:由所选基因构建的FI网络中的基因根据基因评分进行着色。有关如何使用FI网络的特性的更多信息,请参见基因设置/突变分析.当显示网络视图时,您可以打开路径图。要回到以前的网络视图,请关闭所有显示的路径图,然后在左侧控制面板的“网络”选项卡中选择网络。根据黑豹的同源映射文件,从人类路径预测小鼠路径。更多细节,请参阅:在Reactome中的推断事件.利用所提供的映射文件,从人体功能交互网络中预测出鼠标人体功能交互网络MGI,使用此链接下载:http://www.informatics.jax.org/downloads/reports/HOM_MouseHumanSequence.rpt.一个人类基因可以映射到多个老鼠基因上。因此,小鼠FI网络可能显示了一个带有多个小鼠基因注释的节点。例如,请参见下面的内容:原始的人类KLK3被映射到Klk1b9、Klk1b21和其他许多节点,它们都列在底部的节点表中,并在网络视图中作为该节点的标签。目前连接到FI网络中的这些节点指向人类基因(如GeneCard)。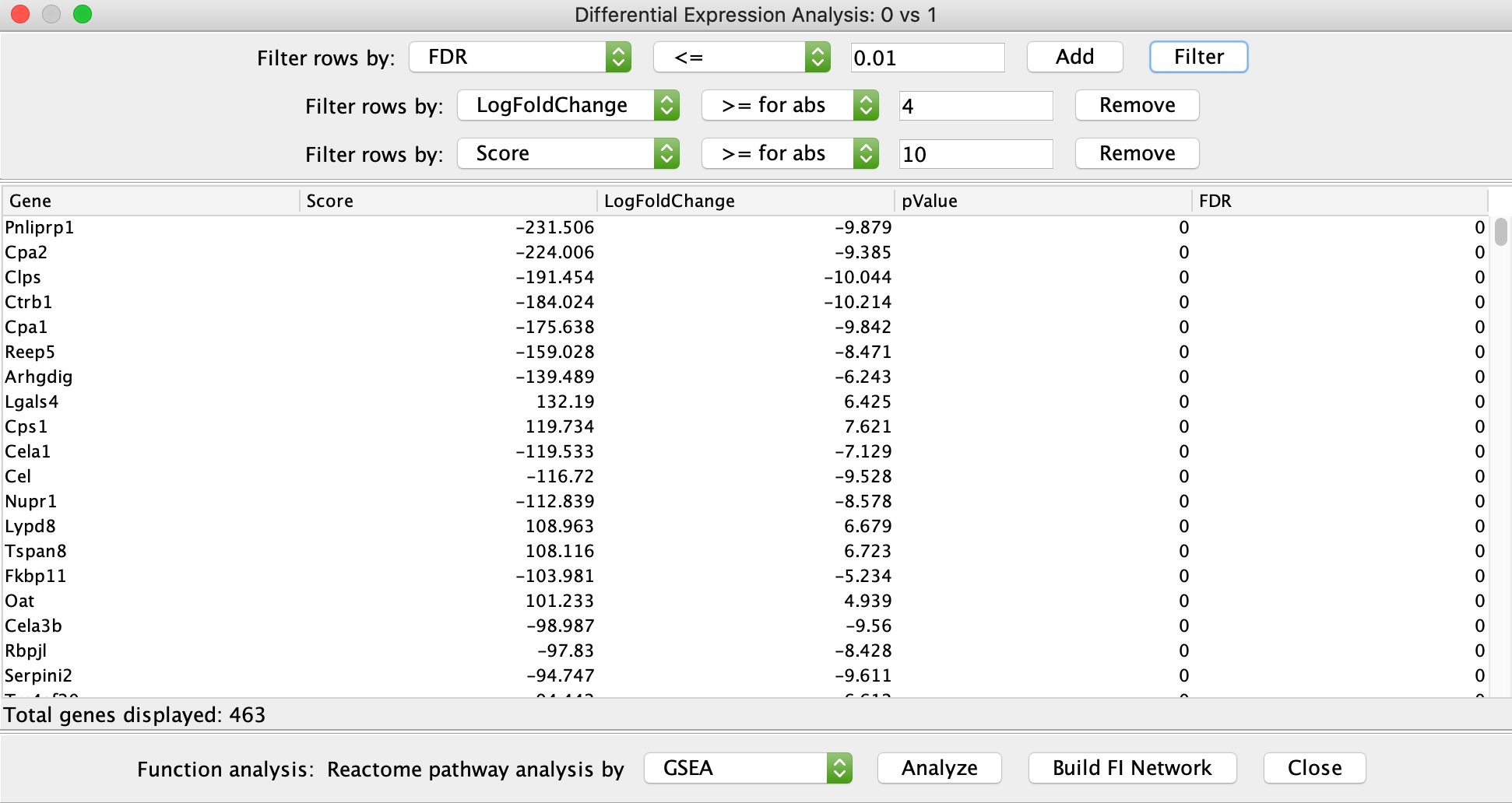 差异表达分析结果
差异表达分析结果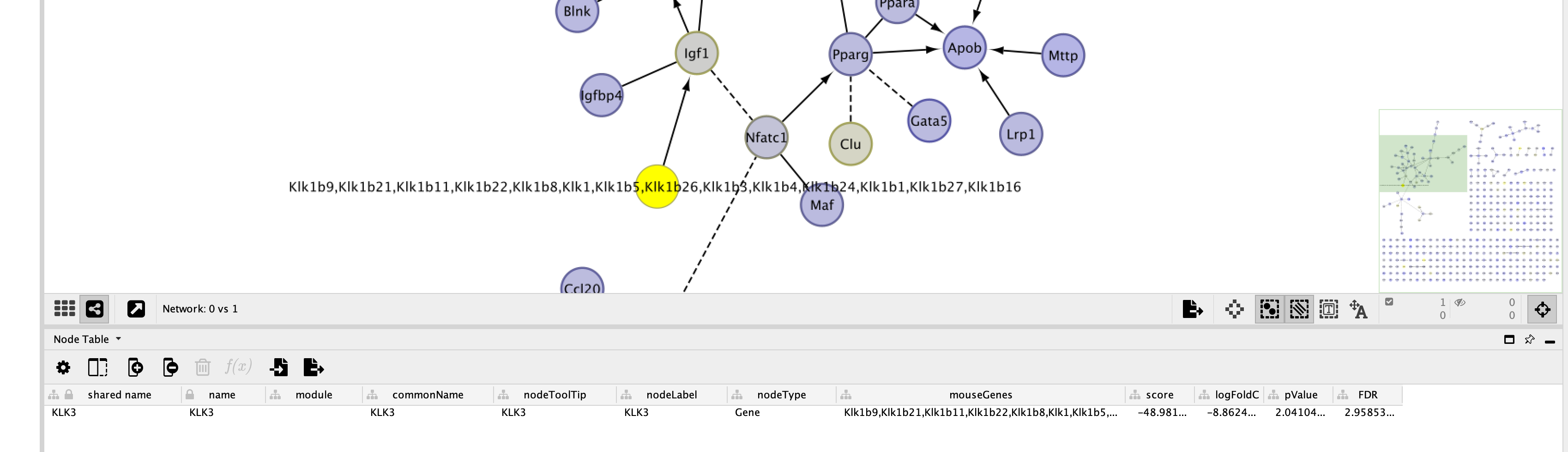 一个人类基因映射到多个小鼠基因
一个人类基因映射到多个小鼠基因 - 建立监管网络:将转录因子(TFS)与其目标之间的底层基因调节网络推断出一个或多个细胞簇之间。这种方法是由秋等人的利用Scribe从耦合单细胞表达动力学推断因果基因调控网络. 然而,ReactomeFIViz提供的当前实施方案使用延时基因共表达来推断TFs与其靶标之间的潜在因果关系,而不是“受限定向信息(RDI)”,并限制了TFs与其靶标之间基于多萝西娅. 要执行此分析,请在以下对话框中设置参数:请注意:您可以选择Spearman、Pearson或Kendal进行基因表达相关性计算。细胞时间类型是潜伏期、速度假时间、细胞追踪或dpt假时间中的一种,它们是在轨迹推断期间计算的细胞属性。前两种类型是在RNA速度分析期间生成的(见下文)。如果ReactomeFIViz无法找到这些变量中的任何一个,您将被要求首先执行dpt_伪时间分析。时间延迟用于进行延迟基因共表达计算。例如,TF的表达为[1,2,3,4,5,6,7,8,9,10],其目标之一的表达为[11,12,13,14,15,16,17,18,19,20]。如果时间延迟=4,则计算[1,2,3,4,5,6]和[15,16,17,18,19,20]之间的相关性。上述对话框中的时间延迟是指最大时间延迟。因此,如果输入7作为时间延迟,则用于相关性计算的实际时间延迟值为[1,2,3,4,5,6,7]。相关性计算7次,并使用这些计算出的相关性值的最大值。对于单元组,您可以选择全部、一个单元群集或多个单元群集(在Mac下按住命令键或在Windows下按住控制键)。
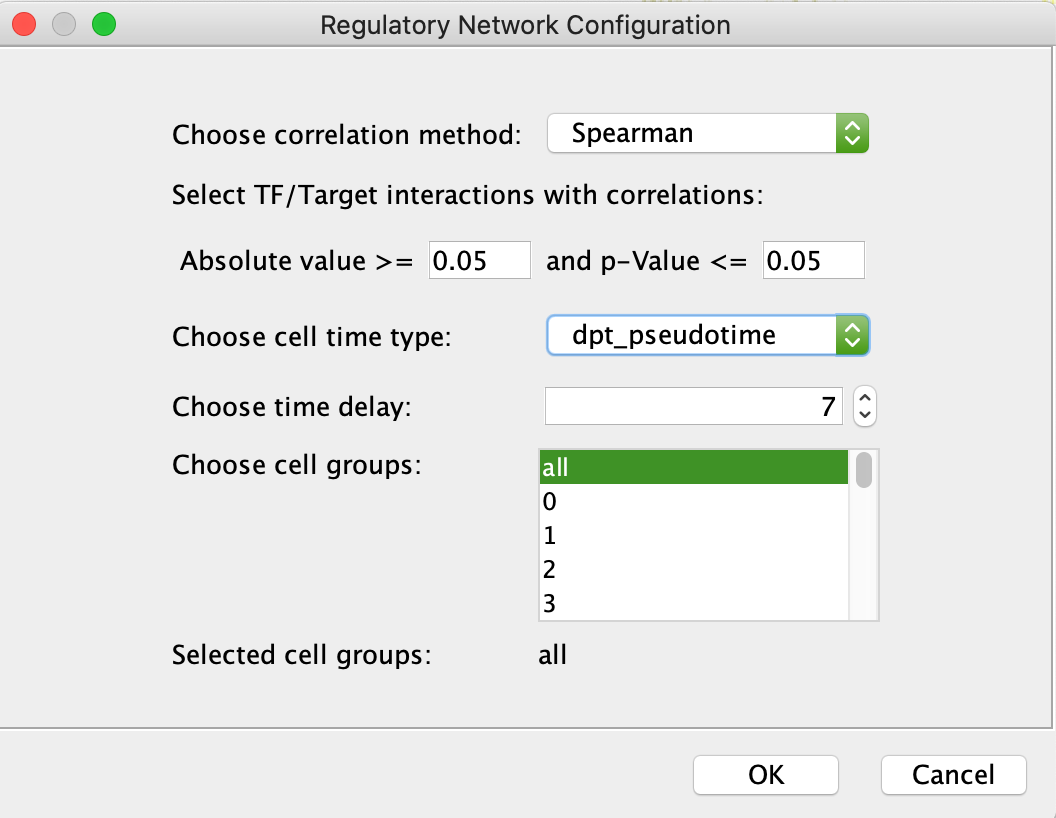 基因调控网络推断装置
基因调控网络推断装置
计算所有TF/靶标相互作用的相关性,然后构建调控网络可能需要几分钟时间。下面的例子展示了最终的基因调控网络是什么样的:请注意:一种称为“调控网络样式”的样式是用来渲染生成的基因调控网络。使用这种风格,tf被渲染为菱形,而他们的目标是圆形。最初的多萝西娅相互作用提供注释:->激活和-|抑制。红色为正相关,蓝色为负相关。计算的相关性可能与多萝西娅的原始注释不匹配。例如,在上图中,一个TF和它的一个靶标Scg3之间的相互作用被注释为抑制。然而,实际计算的斯皮尔曼相关性是正的。边宽与绝对相关值成正比。您可以在Cytoscape中调查这种样式以获得更多信息。 部分基因调控网络
部分基因调控网络 - 项目新数据:将另一个数据集投影到显示的网络上。该特性使用摄取函数在scanpy中集成一个新的数据集到用于生成网络的数据集上,显示新数据集中的单元格可以映射到显示的网络。要执行此分析,请在以下对话框中输入数据文件:请注意:您不能在此对话框中选择方法。预计应使用相同的方法将新数据集投影到现有网络上。投影后,在“投影新数据”菜单下添加一个名为“切换投影数据”的新弹出菜单,用于切换投影单元格的显示。以下两幅图显示了投影新数据集之前(左)和之后(右)的单元网络。如图所示,新数据集中的大多数单元格被投影到中间和右上角区域周围以绿色、鲑鱼色和玫瑰棕色显示的单元格簇上。
 项目新数据配置
项目新数据配置

- 保存分析结果:将所有的分析结果保存到本地文件中h5ad格式。保存的文件可以使用主菜单,Apps/Reactome FI/Single Cell Analysis/Open打开。
- 负载基因表达:将基因的表达值叠加到网络上。点击此菜单后,您可以在输入对话框中输入基因名称。














通过scVelo进行RNA速度分析
RNA速度分析是一种基于未拼接和剪接形式mRNA之间计数比率定量模拟基因mRNA转录动态行为的有效方法(La Manno等人2018).scVelo在Python中提供了原始方法的增强实现。ReactomeFIViz为您提供了scVelo软件包,以便在Cytoscape中使用图形用户界面进行此分析,而无需编写脚本。有关RNA velocity和scVelo的更多信息,请参阅原始scVelo文档:https://scvelo.readthedocs.io.
- 建立分析:在下面的scRNA序列分析操作对话框中选择“通过scVelo进行RNA速度分析”作为方法,并选择其中一种RNA速度模式。要查看对话框中列出的三种模式之间的差异,请参阅原始scVelo文件,通过动力学建模概括瞬态细胞状态的RNA速度.默认值是“随机”。但是,如果要获取更多动态信息,请选择“动态”。对于你可以用动态模型做些什么,看用scVelo进行动态建模.该分析所需的数据文件应使用velocyto或loompy/kallisto管道进行预处理,并包含未拼接和拼接丰度的两个矩阵。有关如何开始的更多信息,请参阅scVelo教程:开始.
 RNA速度分析配置
RNA速度分析配置 - 可视化RNA速度分析结果:根据你选择的模式,可能需要一段时间来进行RNA速度分析。结果与使用scanpy的标准分析结果相同,但单细胞簇网络显示为有向的、加权的网络,其方向与基于的推断轨迹中的时间相对应PAGA单元群之间连接的方法和权重。下图展示了这种加权的、有向的小区集群网络的示例。请注意预期你会从RNA速度分析中看到不同于标准分析的结果。
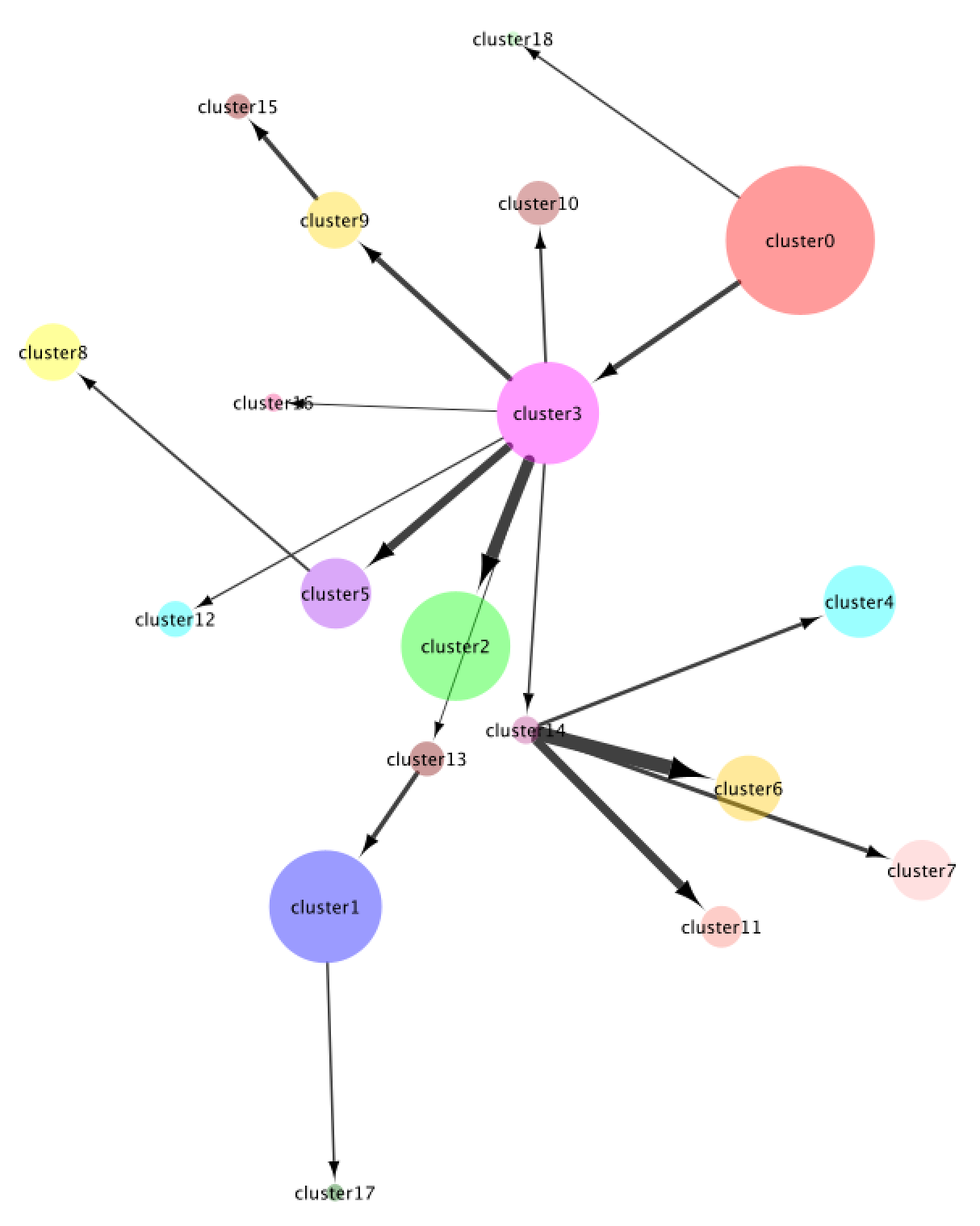 RNA速度集群网络
RNA速度集群网络 - 分析RNA速度结果:通过RNA velocity分析生成的显示网络的大多数分析功能与通过scanpy进行的标准分析的功能相同。但是,RNA速度分析提供的细胞特征比标准分析多得多,如以下弹出菜单所示:请注意要理解这些RNA速度特定细胞特征的含义,请参阅scVelo原版教程:scVelo教程.除了以上RNA速度特定的细胞功能,新增了一个弹出菜单组供您进行一些RNA速度特定的数据分析和可视化,如下所示:
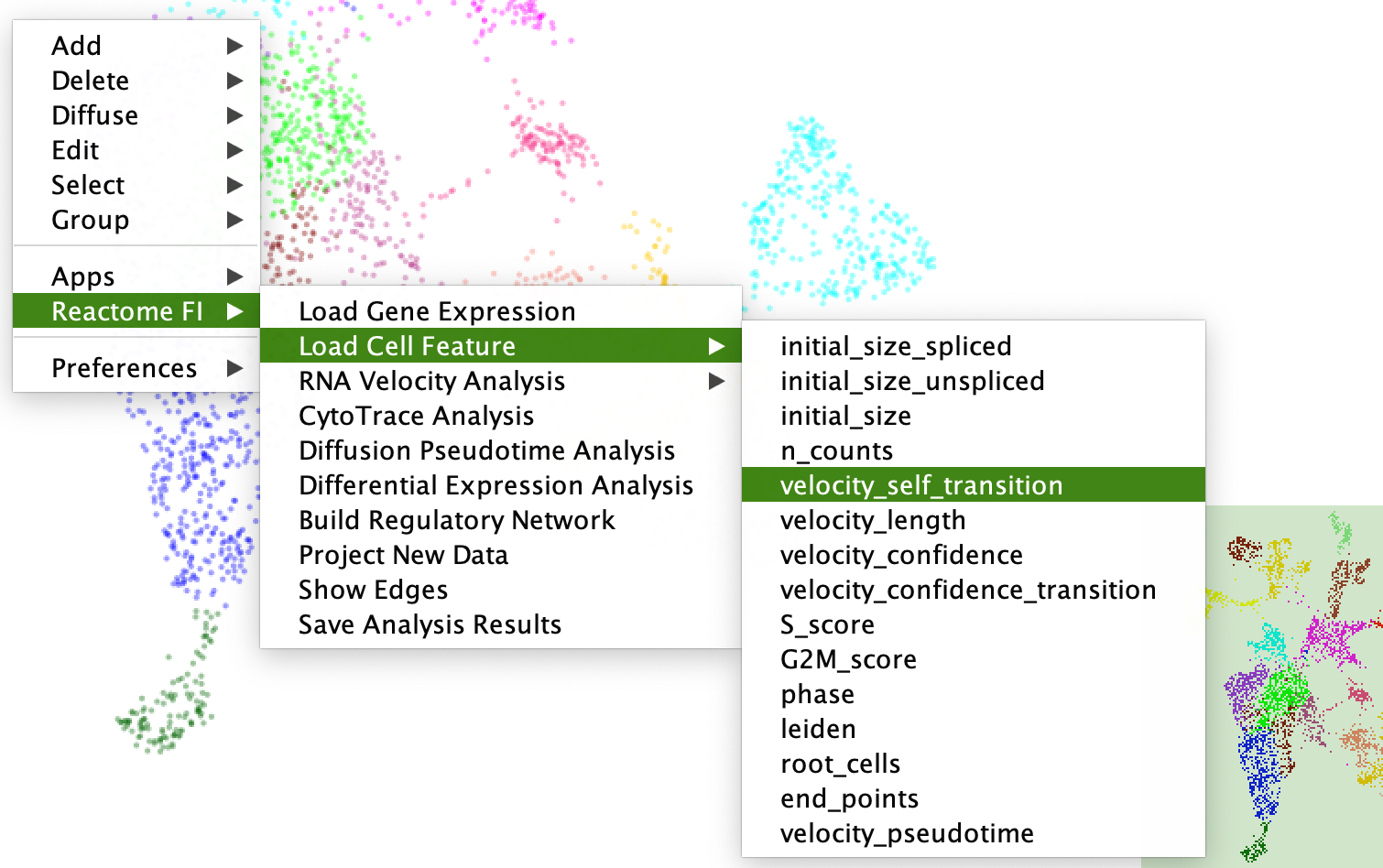 RNA速度细胞功能弹出菜单请注意参考scVelo教程的嵌入,嵌入网格,嵌入流和基因速度:RNA速度基本知识.ReactomeFIViz利用scVelo的可视化特性为这些图形生成图像文件,然后自动打开它们。为了保存这些文件,您可能需要将它们保存到指定的文件中。否则,当您关闭Cytoscape时,它们将被自动删除。
RNA速度细胞功能弹出菜单请注意参考scVelo教程的嵌入,嵌入网格,嵌入流和基因速度:RNA速度基本知识.ReactomeFIViz利用scVelo的可视化特性为这些图形生成图像文件,然后自动打开它们。为了保存这些文件,您可能需要将它们保存到指定的文件中。否则,当您关闭Cytoscape时,它们将被自动删除。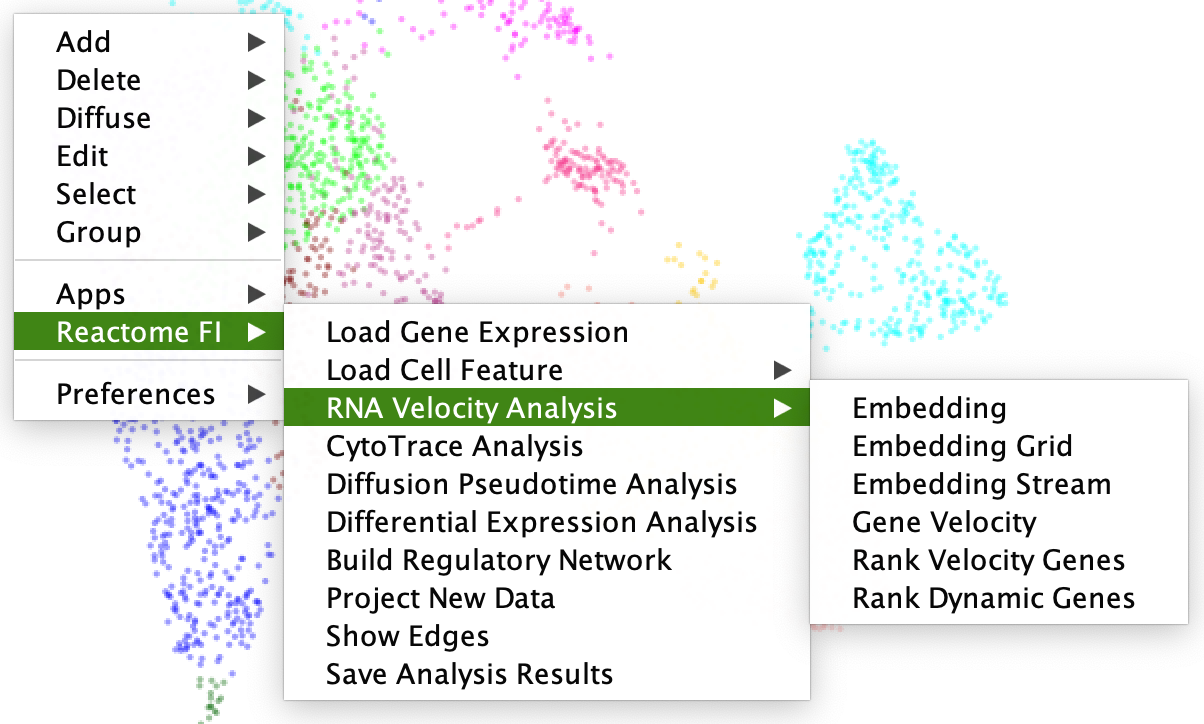 RNA速度弹出菜单
RNA速度弹出菜单- 排名速度基因:使用scVelo的rank_velocity_genes功能,基于差异表达分析对单个细胞簇中的基因进行排序:rank_velocity_genes。此分析返回的单个簇的前250个基因显示在下图所示的表格中。您可以使用二项测试进行路径富集分析,或为选定的细胞簇构建FI网络。请注意:您可以过滤表格中显示的基因。然而,在进行通路富集分析或构建FI网络时,需要使用选定细胞簇的所有250个基因。
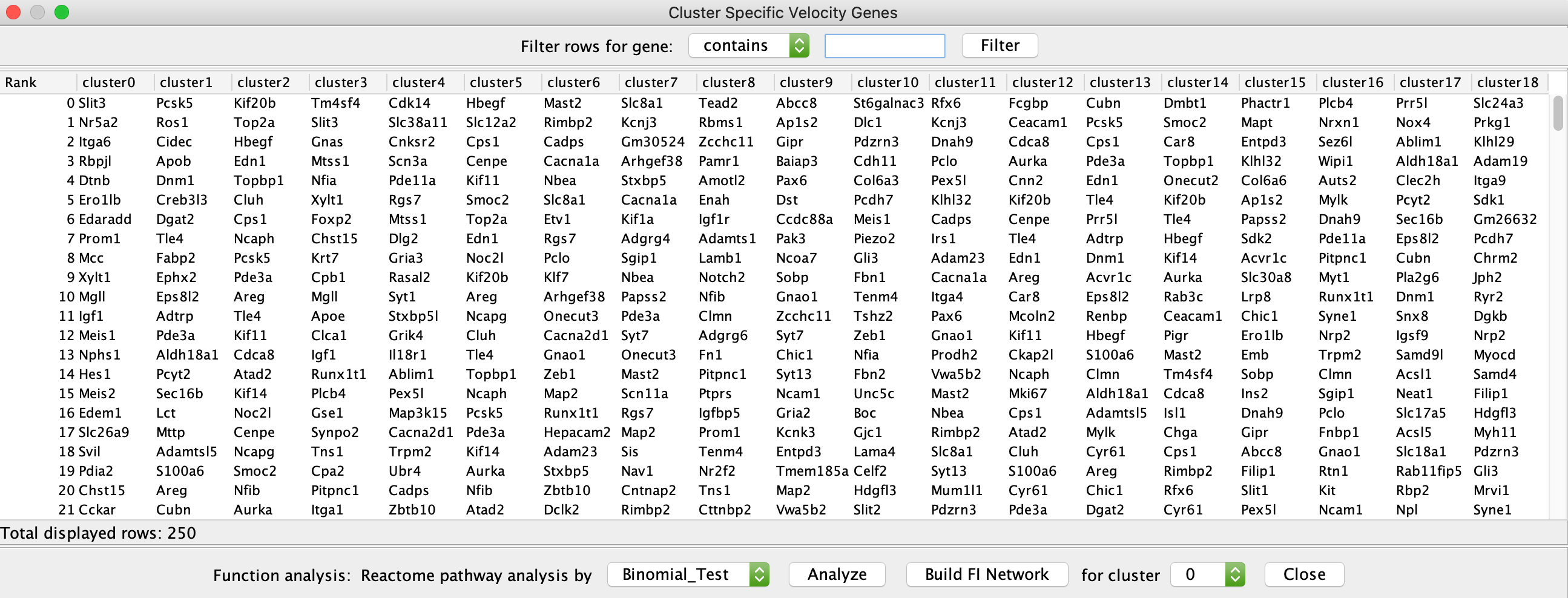 RNA速度Rank基因表
RNA速度Rank基因表 - 等级动态基因:如果你的RNA速度分析选择动态模式,你也可以做“Rank dynamic Genes”。该特性基于scVelo函数,秩动力基因.其输出和功能与“秩速度基因”相同。
- 排名速度基因:使用scVelo的rank_velocity_genes功能,基于差异表达分析对单个细胞簇中的基因进行排序:rank_velocity_genes。此分析返回的单个簇的前250个基因显示在下图所示的表格中。您可以使用二项测试进行路径富集分析,或为选定的细胞簇构建FI网络。





FI网络相关的其他特性
查询FI源
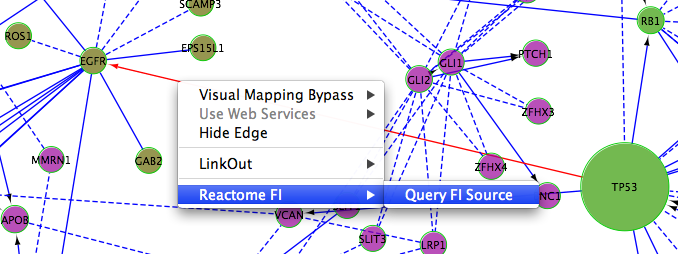
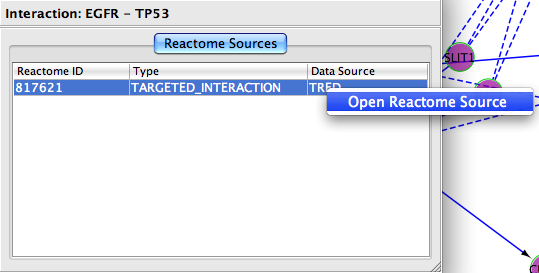
选择一条边,右击它,得到弹出菜单的边。选择一个名为“还原FI/查询FI源”的菜单。如果FI是从经过策划的途径或反应中提取出来的,则会显示原始数据源的对话框。双击显示表格中的一行,显示FI来源的详细网页。如果所选的FI是预测的FI,则应显示该FI的证据。
获取节点的FIs
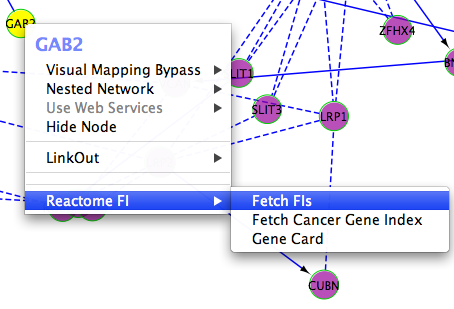
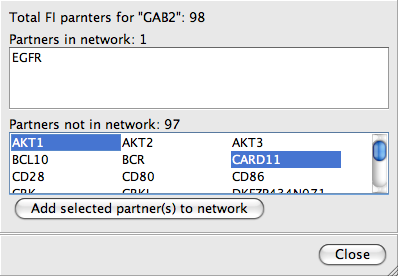
可以查询节点的所有FIs。在网络面板中选择一个节点,然后右击它以获得节点的弹出菜单。选择一个菜单叫做“Reactome FI/Fetch FI”。选中节点的FI伙伴关系将分为两部分显示:网络中已显示的伙伴关系和网络中未显示的伙伴关系。您可以从第二部分中选择合作伙伴来扩展显示的网络。
显示途径图
可以为途径命中率显示路径图。在“网络中的路径”或“模块中的路径”选项卡中选择一个路径,右键单击以获取通路的弹出菜单。从弹出菜单中选择“显示路径图”
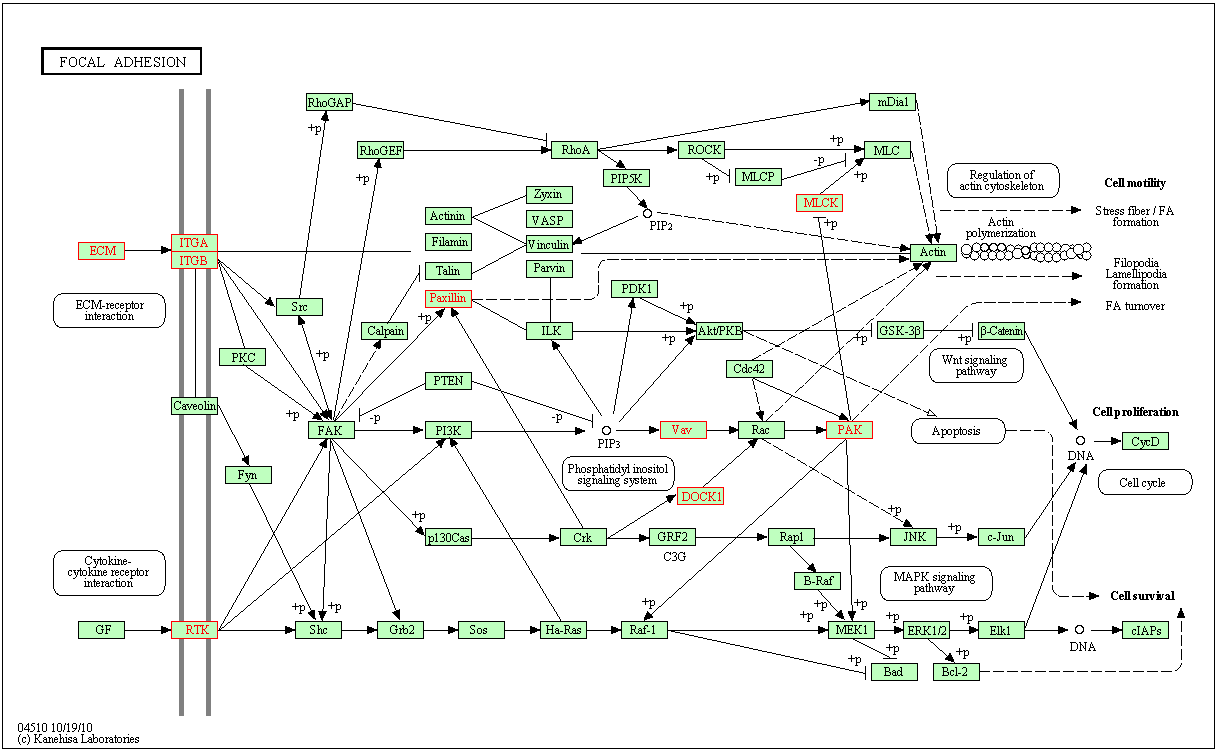
.如果路径是从KEGG导入的,则KEGG路径图页面将在浏览器中显示,节点基因将在“节点”列中以红色突出显示(用于路径图中的文本和边框)。如果路径来自Reactome或其他非kegg数据库,路径图应该显示在一个单独的窗口中。如果路径是由Reactome项目策划的,则应该显示人工布局图(如果有的话)。否则,应该显示自动布局的图表。显示网络中的基因或蛋白质应该用蓝色突出显示。通过使用一个名为“视图实例”的弹出菜单,可以查看图表窗口中显示的节点和反应的详细注释。可以使用窗口底部的缩放滑块放大/缩小显示的图表。图表可以通过右上角的概述窗口进行平移。
载入癌症基因索引注释
Reactome FI插件可加载NCI癌症基因注释索引显示在网络中的基因/蛋白质。有两种方法可以显示这些注释:当没有选择对象时,使用名为“Load Cancer Gene Index”的弹出菜单(左图),对选中的节点使用另一个弹出菜单“Fetch Cancer Gene Index”(右图)。
通过使用第一种方法,用户可以加载NCI疾病术语树,并在左侧面板中显示树。用户可以在树中选择疾病术语,所选疾病的所有基因或蛋白质已被注释,其子术语将被选择。
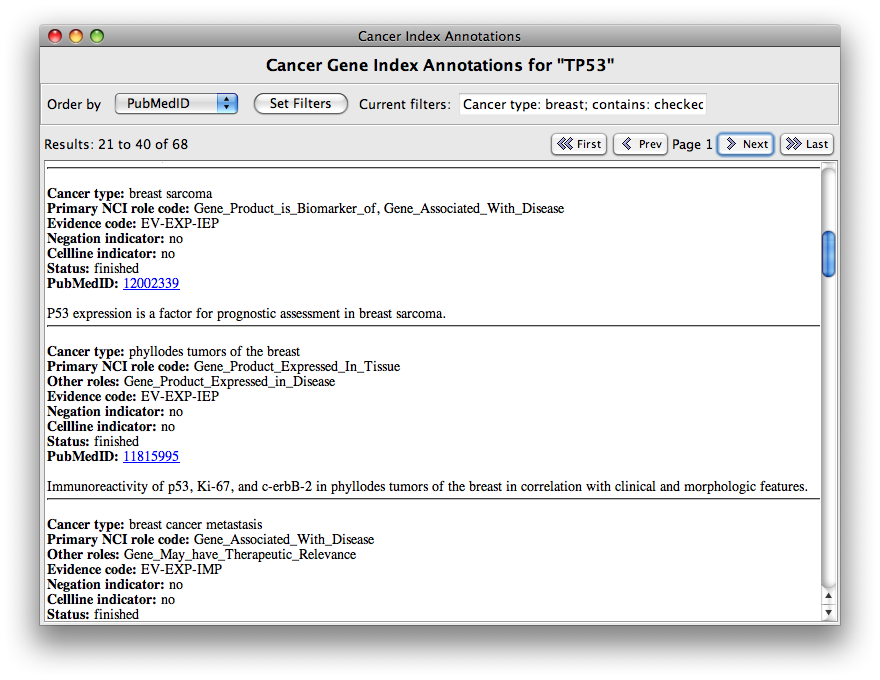
通过使用第二种方法,用户可以查看所选基因或蛋白质的详细注释。用户可以根据PubMedID、癌症类型和注释状态对这些注释进行排序,还可以根据几个标准过滤注释。
生存分析
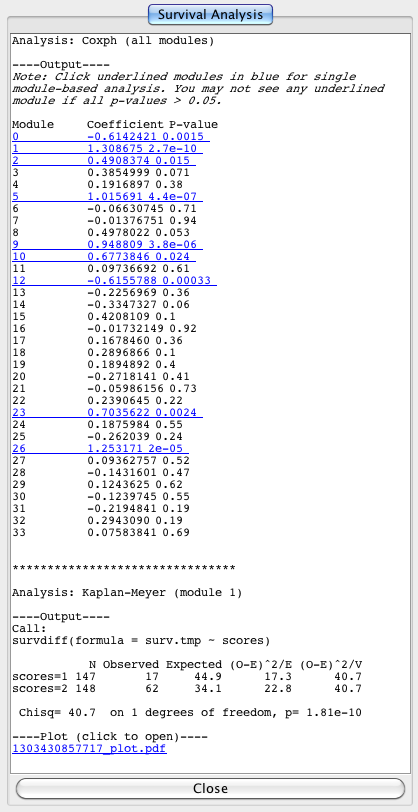
生存分析基于服务器端R脚本,可以进行COXPH或KAPLAN-MEIER生存分析。要进行生存分析,应提供包含至少三列的选项卡分隔的文本文件。三列的名称应该是:样本,软态和病变。例如,请参阅此生存信息文件下载van de Vijver等人的研究:Nejm_Clin_Simple.txt,这已经简化了我们的分析目的。要进行生存分析,请使用弹出菜单“分析模块功能/生存分析......”(见下文)
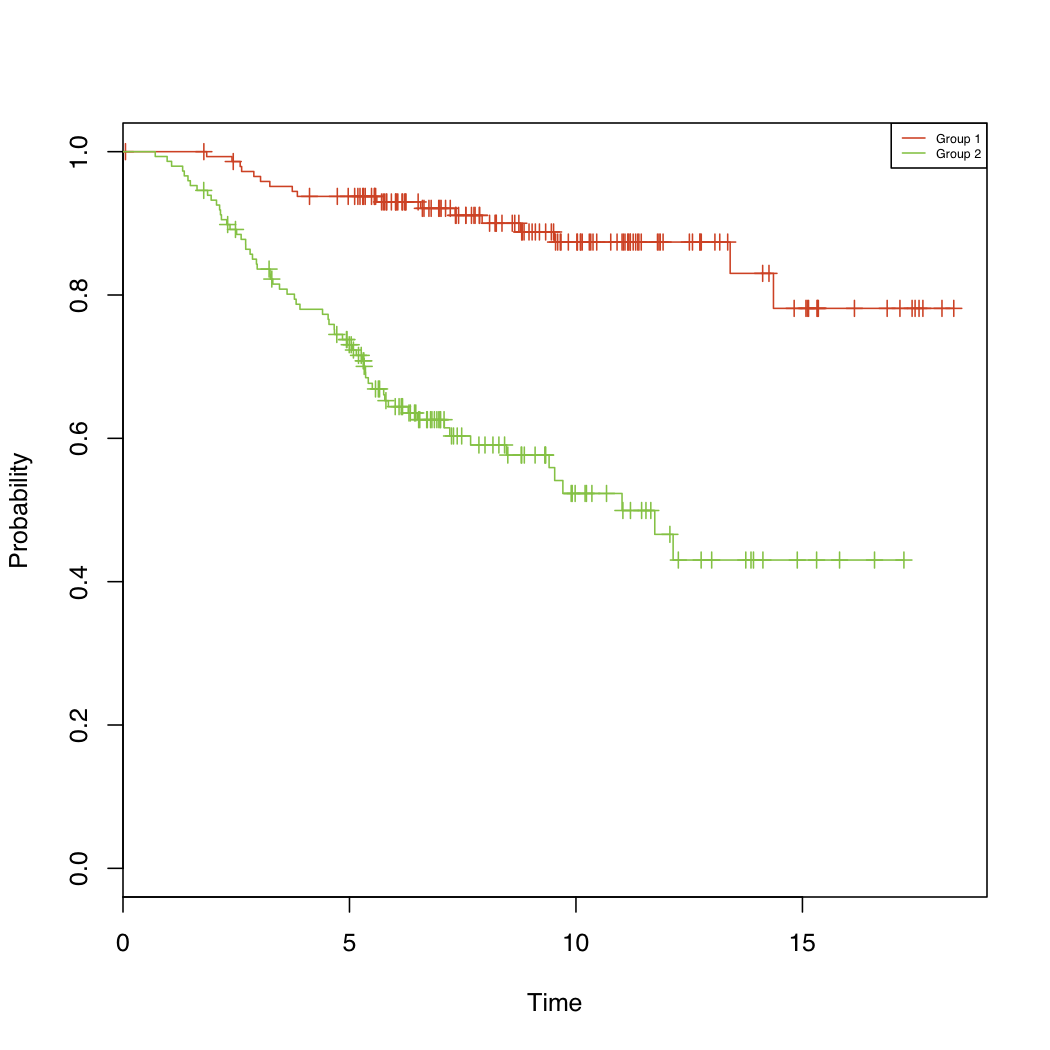
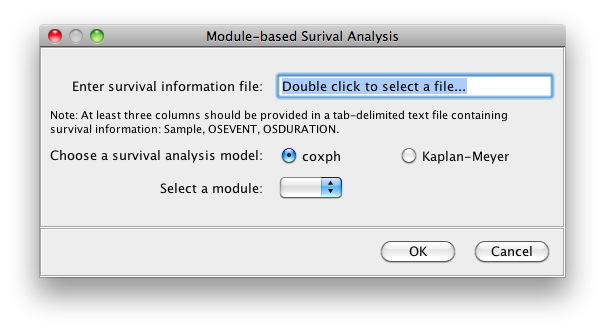
在生存分析对话框(下图)中,双击文本字段,选择包含用于构建所显示的FI子网络的样本生存信息的文件(注意:如果仅使用基因集文件构建所显示的FI子网络,则无法进行生存分析)。你可以选择coxph或Kaplan-Meier模型进行生存分析。如果选择Kaplan-Meier模型,则必须选择一个模块进行分析。在kaplan meier分析中,所有样品将被分为两组:样品没有突变基因在选定的模块(组1)和样品在模块突变基因(组2)。建议先运行coxph模块没有选择任何模块为了看哪个模块最重要相关生存时间。之后,您可以专注于生存分析的一些特定模块。
生存分析的结果将显示在右侧的结果面板中,标签为“生存分析”(左下)。你可以做多个生存分析。服务器端R脚本返回的所有结果将显示在这个面板中,并根据生存分析对话框中的参数选择显示标签。最后一个结果将被选择为默认结果。对于每个分析,结果面板中最多显示三个部分:Output、Error和Plot。如果分析没有返回警告或错误,则错误部分可能不会显示。coxph(所有模块)分析中p值小于0.05的模块的行以蓝色显示,并带有下划线。您可以单击这些模块来进行快速的基于单模块的生存分析,而不需要执行上述步骤。基于单模块的Kaplan-Meier分析将显示一个plot文件。您可以单击该文件以查看实际的图(右下)。 You may need to save the plot file for your future use.